
Abstract
The peptide hormone glucagon, discovered in late 1922, is secreted from pancreatic alpha cells and is an essential regulator of metabolic homeostasis. This review summarises experiences since the discovery of glucagon regarding basic and clinical aspects of this hormone and speculations on the future directions for glucagon biology and glucagon-based therapies. The review was based on the international glucagon conference, entitled ‘A hundred years with glucagon and a hundred more’, held in Copenhagen, Denmark, in November 2022. The scientific and therapeutic focus of glucagon biology has mainly been related to its role in diabetes. In type 1 diabetes, the glucose-raising properties of glucagon have been leveraged to therapeutically restore hypoglycaemia. The hyperglucagonaemia evident in type 2 diabetes has been proposed to contribute to hyperglycaemia, raising questions regarding underlying mechanism and the importance of this in the pathogenesis of diabetes. Mimicry experiments of glucagon signalling have fuelled the development of several pharmacological compounds including glucagon receptor (GCGR) antagonists, GCGR agonists and, more recently, dual and triple receptor agonists combining glucagon and incretin hormone receptor agonism. From these studies and from earlier observations in extreme cases of either glucagon deficiency or excess secretion, the physiological role of glucagon has expanded to also involve hepatic protein and lipid metabolism. The interplay between the pancreas and the liver, known as the liver–alpha cell axis, reflects the importance of glucagon for glucose, amino acid and lipid metabolism. In individuals with diabetes and fatty liver diseases, glucagon’s hepatic actions may be partly impaired resulting in elevated levels of glucagonotropic amino acids, dyslipidaemia and hyperglucagonaemia, reflecting a new, so far largely unexplored pathophysiological phenomenon termed ‘glucagon resistance’. Importantly, the hyperglucagonaemia as part of glucagon resistance may result in increased hepatic glucose production and hyperglycaemia. Emerging glucagon-based therapies show a beneficial impact on weight loss and fatty liver diseases and this has sparked a renewed interest in glucagon biology to enable further pharmacological pursuits.
Graphical Abstract

Similar content being viewed by others


The discovery of glucagon
The discovery of insulin in 1921 by Banting et al [1] was paralleled by the observation that pancreatic extracts [2] and crude insulin preparations [3] induce a brief hyperglycaemic episode before glucose levels decrease below baseline. Trying to optimise insulin purification, Charles Kimball and John Murlin, in 1922, isolated a pancreatic factor that elevates blood glucose in rabbits and dogs [4]. Consistent with its ability to oppose the hypoglycaemic effect of insulin, the factor was called the ‘glucose agonist’ or, for short, ‘glucagon’ [4].
During the following three decades, the hormonal status of glucagon was not generally accepted, but several groups had explored the possible mechanisms underlying the hyperglycaemia induced by glucagon (at that time it was also termed the ‘hyperglycaemic-glycogenolytic factor’ due to its effect on glycogenolysis) [5]. In 1953, glucagon was purified and crystallised [6] and, in 1957, the 29-long amino acid sequence was determined by scientists from the Lilly Research Laboratories [7]. Meanwhile, Christian de Duve’s group and Earl Sutherland et al localised the secretory origin of glucagon to the pancreatic alpha cells and also identified the gastric mucosa as a source of glucagon [8, 9]. Finally, with the introduction of the first glucagon radioimmunoassay by Roger Unger and colleagues in 1959 [10], it became possible to measure plasma concentrations of glucagon allowing physiological studies of glucagon and its role in pathophysiological processes. Subsequently, it was discovered that individuals with diabetes had increased plasma glucagon concentrations [11]. A brief historical overview of glucagon biology is shown in Fig. 1.

Key historical discoveries of glucagon biology over the last 100 years. Pancreatic alpha cells (shown in the schematic) are the source of glucagon and the major focus in glucagon biology and research. Glucagon, blue triangles; glucose, orange circles. Created with BioRender.com. This figure is available as part of a downloadable slideset
A key focus of glucagon research has been to understand the regulatory mechanism(s) controlling its secretion. In the next section, therefore, we introduce and discuss the most common aspects of alpha cell glucagon secretion.
Regulation of glucagon secretion
Introduction to the area
Major contributions to our understanding of the regulation of glucagon secretion were made in the Unger laboratory. Using a variety of experimental models, glucose sensing and amino acid signalling in pancreatic islets were shown to play a fundamental role in glucagon secretion [12]. Unger and co-workers mapped secretory responses to meal ingestion and also discovered the absolute or relative hyperglucagonaemia in people with type 2 diabetes [13]. However, it soon became clear that glucagon-like substances from the gut, as identified by Sutherland and de Duve [14], could interfere with the measurement of glucagon. Furthermore, from emerging evidence that gastrointestinal ‘glucagon’ was functionally different from pancreatic glucagon and secreted in response to different physiological stimuli, it was realised that measuring glucagon was not simple. Moreover, low concentrations and the inherent instability of glucagon further complicated the interpretation of data. It is, therefore, necessary to highlight the methodological and technological advances that have enabled reliable and accurate measurements of glucagon concentrations in plasma.
Glucagon assays
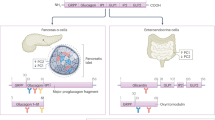
A consideration of the biosynthesis of the glucagon molecule illustrates the complexity of glucagon measurements. At least five glucagon-like molecules (and perhaps more as a consequence of incomplete or additional protein cleavage) are circulating in the body (Fig. 2) and this puts great demands on the selectivity of glucagon assays [15].

Accurate measurement of glucagon. The precursor of glucagon, termed ‘proglucagon’ (160 amino acids) is expressed in alpha cells and l-cells in the pancreas and intestine, respectively. Expression of proglucagon is also reported in brain tissue and its processing pattern in the brain seems to be similar to that within the intestine. Proglucagon is broken down into several peptides, including glicentin-related polypeptide (GRPP), the major proglucagon fragment and glucagon. In the pancreas, glucagon is formed and also detectable amounts of glucagon 1-61. In intestinal l-cells, proglucagon is processed to form glicentin and oxyntomodulin. All of these molecules share amino acid sequences with glucagon, complicating the process of measuring glucagon using antibody-based approaches. For example, N-terminal glucagon antibodies also detect oxyntomodulin, while antibodies that target the mid portion of glucagon (‘side-viewing antibodies’) also detect glicentin, oxyntomodulin and glucagon 1-61. C-terminal glucagon antibodies would also detect glucagon 1-61. Consequently, sandwich ELISAs are pertinent to accurate measurements of glucagon. GLP-1 and GLP-2 are not shown but these molecules also have sequence similarities with glucagon. Created with BioRender.com. This figure is available as part of a downloadable slideset
The famous antibody 30K, used since 1975 in Unger’s and many other laboratories [16, 17], is directed against the C-terminus of glucagon and, therefore, does not show major cross-reactivity with the gut-derived products of the glucagon gene (only the glucagon 1-61 peptide from the pancreas can interfere with C-terminal-targeting assays, but this circulates in low concentrations and is secreted in parallel with pancreatic glucagon [18]). C-terminal (so-called ‘specific’) antisera have been used by several laboratories [19]. Antibodies directed against the mid portion of glucagon (‘cross-reacting’ antisera) not only react with glucagon but also with other proglucagon by-products, glicentin, glucagon 1-61 and oxyntomodulin, and thus are of limited use. N-terminal antisera are also of limited value due to glucagon and oxyntomodulin cross-reactivity (see Fig. 2). Apart from the specificity-related problems, glucagon assays must be able to detect subtle changes in glucagon levels (as little as 1 pmol/l) to identify glucose-mediated suppression of secretion. Sandwich ELISA technology, when strictly based on N- and C-terminal-binding antibodies, can solve these problems [20]. Although such assays are now available, their specificity clearly depends on the exact binding epitopes; even the slightest aberration from terminal reactivity of the antibodies will negatively affect the accuracy of the assay. Moreover, physiological context can sometimes challenge assay performance. For example, after Roux-en-Y gastric bypass, the extreme mass of glicentin and other proglucagon-derived peptides secreted after eating can cause interference even when using well-characterised sandwich ELISAs [21]. Detailed guidelines for the measurement of glucagon are discussed elsewhere [15].
We now move on to describe and discuss the further complexities underlying glucagon secretion.
Alpha cell secretion
The regulation of glucagon secretion by paracrine, hormonal and nutrient stimuli is complex (Fig. 3) and still debated [22]. Several factors, such as the experimental setup used (e.g., in vitro vs in vivo), the technique of glucagon measurements (see above) and dosing of the stimuli given (i.e. physiological vs pharmacological levels) may influence results from studies in this area. An important impetus to the study of alpha cell secretion of glucagon is the notion that hyperglucagonaemia and the apparently abnormal regulation of glucagon by glucose in individuals with type 2 diabetes is due to alpha cell dysfunction [23]. Furthermore, the up to eightfold increase in glucagon secretion that is normally seen in response to hypoglycaemia [24] is often impaired in individuals with type 1 diabetes. It has also been suggested that the intrinsic control of glucagon secretion in response to high glucose is impaired during the development of diabetes [25]. On the other hand, most observations suggest that alpha cells in individuals with obesity and type 2 diabetes do exactly what they are supposed to do: glucose inhibits glucagon secretion (although this may be delayed [26] and/or depend on the route of glucose administration [27]), while proteins/amino acids stimulate secretion [28]. Instead, it has been suggested that the basal and postprandial hypersecretion of glucagon in type 2 diabetes could be owing to elevated plasma amino acid levels and gastrointestinal factors [29]. One of the reasons for the disparity in findings is the lack of high-quality alpha cell models. Alpha cells are difficult to isolate and respond poorly in isolation, and reliable cell lines do not exist.

Regulation of glucagon secretion. Glucagon secretion is controlled by numerous mechanisms, with only a few selected mechanisms being shown in the figure. Intra-islet communication by delta and beta cells may suppress glucagon release in response to secreted factors. The role of insulin in suppressing glucagon secretion is debated. In contrast, somatostatin exerts a well-characterised inhibitory effect on glucagon secretion by alpha cells through somatostatin subtype receptors (SSTRs). Nutrients, such as glucose, fatty acids and amino acids play a major role in the regulation of glucagon secretion. Additionally, both neural and extrapancreatic factors, such as peptides secreted from the gastrointestinal tract (e.g. GLP-1, GIP and oxyntomodulin), may couple nutrient absorption to the regulation of glucagon release. Several drugs, such as GLP-1 analogues, sodium−glucose cotransporter (SGLT) inhibitors and neprilysin inhibitors may also modulate alpha cell function by direct and indirect mechanisms (not shown). GLP-1-based agents are glucagonostatic, exerting their effects by stimulating somatostatin release. SGLT inhibitors potentially have both direct (via the SGLT1 transporter in alpha cells) and indirect (by lowering blood glucose through blocking renal reabsorption of glucose) effects. Neprilysin inhibitors may elevate plasma glucagon levels by directly protecting endogenous glucagon from degradation. Inhibitors of glucagon secretion shown in red text; stimulators of glucagon secretion shown in blue text. FFAR, free fatty acid receptor; GLP1-R, GLP-1 receptor; INSR, insulin receptor; PLC, phospholipase C. This figure is available as part of a downloadable slideset
In in vivo studies, in the fasting state active secretion of glucagon is evident and leads to peripheral plasma concentrations of around 10 pmol/l (2–3-fold higher in the portal vein), depending on the glucagon assay. Normal alpha cell function results in suppression of glucagon concentrations to very low levels (~1 pmol/l) during bouts of elevated plasma glucose (e.g. during i.v. glucose infusion) or, conversely, stimulation of glucagon secretion with low glucose levels, as is the case in insulin-induced hypoglycaemia [30]. Thus, glucose plays a fundamental role for glucagon secretion, yet the mechanisms involved in this phenomenon are unclear, including glucose sensing by the alpha cell. Conventional knowledge suggests that alpha cells take up glucose via the glucose transporter GLUT1, and perhaps also via sodium-dependent glucose cotransporters (sodium−glucose cotransporter [SGLT] 1 and 2); however, expression levels of SGLTs are low in alpha cells and inhibitors of these transporters may not influence glucagon secretion [31]. It may be questioned whether the alpha cells depend on glucose for energy supply at all. Any glucose that does enter the alpha cell is assumed to be metabolised (via glucokinase, glycolysis and the Krebs cycle) to produce ATP, which interacts with ATP-sensitive potassium channels (KATP channels), leading to cell depolarisation and generation of action potentials [32, 33]. However, despite evidence that glucose promotes alpha cell depolarisation, counter-intuitively, low glucose stimulates glucagon secretion and high glucose inhibits it [34]. There is currently no satisfactory explanation for this, and the lack of effects of sodium channel blockers (which prevent action potentials) on glucagon secretion also contradicts this. Instead, paracrine interactions of alpha cells with neighbouring somatostatin-secreting delta cells may partly explain glucose-mediated regulation of glucagon secretion, as discussed in more detail below [35].
Fatty acids or amino acids may be more important than glucose for regulating glucagon secretion. The stimulatory effect of certain amino acids may even override the influence of prevailing glucose concentrations [36]. Thus, the alpha cell may depend on amino acids for basal metabolism and secretion. Many amino acids stimulate glucagon secretion [37] and provide the fuels for ATP production. Fatty acids may have a similar function, although deep understanding is lacking [38]. Amino acids play a dominating role in alpha cell secretion, although their transduction mechanisms are poorly understood [39]. Arginine has been known to provide a strong stimulus to glucagon secretion for many years and is often used as a test for alpha cell function [12]. Alanine also provides a potent stimulus to alpha cells [40]. In a systematic search for glucagonotropic amino acids from isolated perfused pancreas preparations, these two amino acids, as well as glycine and proline, stood out as being the most important stimulatory amino acids [41]. The steatotic liver is resistant to the effects of glucagon on amino acid metabolism and, because of this, steatosis is associated with elevated levels of most amino acids, which in turn cause hypersecretion of glucagon [42]. Importantly, steatosis does not seem to cause a parallel resistance to the hyperglycaemic effects of glucagon (with glycogenolysis taking place mainly in the central part of the liver acinus and amino acid metabolism taking part in the portal end), so hyperglucagonaemia still contributes to diabetes-associated hyperglycaemia [42, 43]. Unfortunately, there are huge gaps in our knowledge regarding the effects of individual amino acids on glucagon secretion. Randomised crossover studies in healthy individuals using infusion of labelled individual amino acids combined with isotope tracing and accurate measurements of glucagon are required.
Intra-islet regulation of glucagon secretion
Glucagon secretion is important for insulin secretion and vice versa. Stimulation of beta cells via glucagon binding to the glucagon receptor (GCGR) as well as the glucagon-like peptide 1 (GLP-1) receptors is essential for amino acid-induced insulin secretion. Similarly, alpha cells are dependent on beta cells and somatostatin-producing delta cells for appropriate functioning [44, 45]. Alpha cells are exquisitely sensitive to inhibition by somatostatin, which is capable of halting glucagon secretion [35]. Conversely, blockade of somatostatin receptors (a combination of inhibitors of somatostatin receptors 2 and 3 may be required for this) substantially increases glucagon secretion, seemingly independent of glucose levels [46]. This suggests that somatostatin may be involved in glucose-induced inhibition of glucagon secretion, particularly since somatostatin is secreted in response to high glucose levels (>7 mmol/l) [47]. Nonetheless, the role of the beta cells in glucagon secretion is controversial and the mechanisms involved are unclear [48]. In many studies of isolated pancreases, inhibition of glucagon secretion cannot be elicited with perfusate insulin concentrations as high as 1 mmol/l and the potent insulin receptor antagonist S961 also has no effect on glucagon secretion at 1 mmol/l [49]. One may also ask how activation of the insulin receptor, a tyrosine kinase, would bring about acute suppression of glucagon secretion. This is unfortunately unexplored. The immediate conclusion is that beta cells do not influence glucagon secretion via insulin [50]; however, beta cells release more than insulin, including peptides such as urocortin 3 and amylin, with urocortin 3 stimulating somatostatin secretion from delta cells, and it may be through such peptides that the beta cells mediate their glucagonostatic effects [51].
Influence of extra-islet factors
Neural and endocrine factors also influence glucagon secretion. The alpha cells express receptors for a plethora of neurotransmitters and modulators [52]. In pigs, when both the splanchnic and vagal nerves to the pancreas were directly stimulated, both divisions of the autonomic nervous system strongly stimulated glucagon secretion depending on the prevailing plasma glucose concentrations [53]. Regarding the vagal influence, signalling is interrupted in the intrapancreatic ganglia and transmitted, via nicotinic receptors, to secondary neurons innervating the islets. These secondary neurons may also be cholinergic, but non-cholinergic non-adrenergic signalling may also be involved. In particular, vasoactive intestinal polypeptide and pituitary adenylate cyclase-activating polypeptide may be engaged as transmitters [54]. Again, the final outcome is likely to be modulated by paracrine influences (e.g. involving delta cells). The sympathetic nervous system also plays an important role, with direct stimulation of sympathetic fibres increasing glucagon secretion and simultaneous release of adrenaline (epinephrine) from the adrenal medulla also potentially contributing. Inhibitory activity may also be elicited upon alpha-1 adrenergic activity; however, during diffuse sympathetic activity (and also during cold exposure), stimulation of glucagon secretion is to be expected and, because of this, glucagon may be considered a ‘stress hormone’ [55]. This may be particularly important during acute bouts of strenuous exercise, whereas exercise at intermediate intensity appears to stimulate glucagon secretion mainly via slight reductions in plasma glucose levels [56]. There is a general belief that stimulated glucagon secretion only occurs below a certain (low) threshold of plasma glucose, but this only applies to insulin-induced hypoglycaemia. During exercise (where insulin is suppressed) and in other situations where glucose falls in an insulin-independent manner, glucagon secretion increases at the slightest lowering of plasma glucose [57]. Hypoglycaemia will lead to increases in both vagal and sympathetic activity and, evidently, both may participate in the glucagon response to hypoglycaemia in humans. Remarkably, however, it is still not understood why the glucagon response is deficient or even absent in individuals with type 1 diabetes (and long-standing type 2 diabetes [58]), where this deficient response appears to be independent of the presence or absence of autonomic neuropathy [59]. In people with insulin-induced hypoglycaemia, part of the glucagon response appears to be dependent on the modulation of neural input via the brain [60]. The response to meals and amino acids is unimpaired in such individuals.
Extrapancreatic hormones
Obviously, any hormone with an effect on plasma glucose may potentially affect glucagon secretion. Direct effects may be exerted by glucose-dependent insulinotropic polypeptide (GIP) and GLP-1 from the gastrointestinal tract, which stimulate and inhibit glucagon secretion, respectively [61]. GLP-1-induced inhibition of glucagon secretion has been calculated to be responsible for about half of the glucose-lowering effects of exogenous GLP-1 in individuals with type 2 diabetes [62]. The impaired GLP-1 response observed in obesity and type 2 diabetes has, therefore, been implicated in the pathogenesis of hyperglucagonaemia [63]. Remarkably, however, GLP-1 does not inhibit glucagon secretion during insulin-induced hypoglycaemia, whether in individuals with type 2 or type 1 diabetes or control participants [64]; this is, of course, important since it demonstrates that GLP-1-based therapy does not impair this important counter-regulatory mechanism. Indeed, in isolated alpha cells, GLP-1 has been reported to stimulate glucagon secretion, consistent with the expression of a small number of GLP-1 receptors on these cells [65]. One theory is that the inhibition by GLP-1 is normally exerted indirectly via paracrine somatostatin secretion, which markedly diminishes during hypoglycaemia [66]. During hyperglycaemia, the inhibitory mechanism predominates, so the weak stimulatory effect of GIP is mainly visible at lower glucose levels [30]. The actions of GLP-1 on alpha cells may, therefore, primarily be dependent on indirect mechanism involving somatostatin and its receptor located on alpha cells [67].
Moving forward from how glucagon secretion is controlled, we now introduce key aspects of its actions and the inherent experimental challenges in differentiating between the physiological and pharmacological actions of glucagon.
Actions of glucagon
Introduction to the many actions of glucagon
The physiological actions of glucagon are difficult to define due to differences between species (such as the lipolytic effects observed in rats but not in humans [68]), the rapid clearance of glucagon, dose-dependent interaction with the GLP-1 receptor [45], and direct vs indirect actions mediated by activation of the sympathetic nervous system or through secretion of hepatokines, such as fibroblast growth factor 21 (FGF21 [69]). Glucagon possesses effects on multiple organs including the liver, pancreas, kidney, heart and brain (see Fig. 4) [22, 70]. For some of these organs, in particular the brain and heart, its actions are not well established, whereas for the liver, its role in glucose production has been extensively studied. For glucagon to have a direct effect, the GCGR must be expressed but, as mentioned previously, indirect actions may also occur via the GLP-1 receptor [71], promotion of the secretion of hormones (i.e. insulin and FGF21) [45] or activation of the sympathetic nervous system [72]. Such actions, which are important from a pharmacological perspective, are less relevant when discussing the role of endogenous glucagon.

Glucagon action and signalling. Glucagon acts via the GCGR, and also via the GLP-1 receptor when administered at pharmacological doses (not shown). The glucagon/GCGR signalling pathway in hepatocytes is shown, as well as the processes affected by glucagon in selected organs. The exact localisation of the GCGR in the kidney, the intestine and the brain is controversial, while, in the heart, it is debated whether GCGR is expressed at all. Glucagon may directly or indirectly increase heart rate [123], renal excretion of electrolytes [124] and gastric emptying [125]/peristalsis [126]. In contrast, the actions of glucagon on the liver and the pancreas are well established. The image depicts GCGR signalling through G-protein-coupled activation of Gαsubunit and Gq, resulting in cAMP production and Ca2+ signalling, respectively. The non-transcriptional mechanism(s) induced by glucagon are largely unexplored but may include phosphorylation and acetylation of proteins, such as those involved in the urea cycle or glycogenolysis. The transcriptional effects of glucagon on the urea cycle are used an example of a glucagon-induced transcriptional mechanism in the schematic. Glucagon signalling results in an increase in plasma glucose and decreased plasma levels of amino acids and lipids. Created with BioRender.com. This figure is available as part of a downloadable slideset
In the following, we discuss selected physiological features of glucagon.
Hepatic actions of glucagon on glucose production
Glucagon increases blood glucose by enhancing two central hepatic biochemical processes, glycogenolysis and gluconeogenesis. The effects on hepatic glucose production are short-lasting, at least partly because of concomitant glucagon-induced insulin secretion. Also neural or hormonal signals may contribute to the evanescence of glucose production [73]. The hyperglycaemic effect of glucagon may depend both on glycogenolysis in a dose-dependent manner, mediated through cAMP and on transcription-dependent changes in rate-limiting gluconeogenic enzymes, such as PEPCK. Given the absence of GCGRs on muscle cells or adipocytes in humans, the effect of glucagon on gluconeogenesis is likely to depend on substrate delivery, mediated by lipolysis and/or proteolysis. The stabilising effect of glucagon on blood glucose levels during prolonged fasting may depend on neural activity and cortisol, which boost lipolysis and proteolysis. It is likely that the physiological actions of glucagon differ between the fasting and postprandial state and, for the latter, the macronutrient composition of the meal. A high carbohydrate meal would be expected to inhibit glucagon secretion, whereas a mixed meal containing proteins increases glucagon secretion, thus counterbalancing amino acid-induced secretion of insulin, which would otherwise potentially result in hypoglycaemia.
It has been speculated that glucagon also enhances gluconeogenesis in the gut and kidney as both organs possess GCGRs [74] and are believed to generate glucose through processes similar to those in the liver. However, direct evidence for this is lacking.
Hepatic actions of glucagon on amino acid catabolism
Metabolism of amino acids may generate ammonia, which, in the liver, is converted to water-soluble urea through a series of enzymatic reactions, collectively termed ‘ureagenesis’. Glucagon has both transcriptional and non-transcriptional effects on ureagenesis and also increases amino acid uptake through transcriptional upregulation of amino acid transporters [75, 76]. The increase in plasma glucagon levels during a protein-rich meal thereby promotes catabolic utilisation of amino acids in the liver [77]. The exact mode of action of glucagon on ureagenesis is still largely unknown and is discussed elsewhere, in detail [42]. Collectively, these hepatic actions of glucagon contribute to protein metabolism through the liver–alpha cell axis.
The liver–alpha cell axis
The first notions on the liver–alpha cell axis were based on the observation that selective impairment of hepatic glucagon signalling causes dramatic increases in glucagon secretion (hyperglucagonaemia) and also, in animal models, alpha cell hyperplasia [39]. Subsequently, the link was identified as hyperaminoacidaemia [78]. A series of animal and human data from different groups support that glucagon acutely and chronically regulates hepatic amino acid catabolism, while amino acids control glucagon secretion in a relatively glucose-independent manner [36]. The liver–alpha cell axis also involves glucose (as already discussed) and possibly also lipids.
Effects of glucagon on lipid metabolism
Glucagon stimulates hepatic lipolysis and enhances clearance of cholesterol from the circulation [79]. The first indications that glucagon affects cholesterol metabolism date back to the work of Raymond Caren and Lucille Carbo, who in 1956 showed that degranulation of alpha cells by i.v. infusion of cobalt chloride increases circulating levels of cholesterol in rabbits [80]. Supplementation of glucagon was subsequently shown to decrease plasma lipid levels in dogs made hyperlipidaemic by partial pancreatectomy, followed by a series of studies showing that glucagon decreases circulating levels of cholesterol in humans, rodents and dogs [79]. The clearance of cholesterol from the circulation is, at least in part, attributed to glucagon’s ability to increase activity of the LDL receptor and, hence, to promote uptake and clearance of cholesterol via the liver [81]. Glucagon further controls lipid metabolism through inhibition of de novo lipogenesis and acceleration of lipolysis in the liver and the adipose tissue [79]. The lipolytic effect of glucagon is attributed to its ability to enhance activity of hormone-sensitive lipase, the rate-limiting enzyme responsible for breakdown of triglycerides (also termed ‘triacylglycerol’ for intracellularly stored lipids) [82], and to stimulate the secretion of lipolytic hormones, such as growth hormone, cortisol and adrenaline. Signs of glucagon resistance towards hepatic lipid metabolism have been reported in individuals with non-alcoholic fatty liver disease (NAFLD) [83]. Glucagon-mediated elevation of NEFAs increases ketogenesis, and suppression of glucagon secretion using somatostatin may prevent/delay the development of ketoacidosis in individuals with type 1 diabetes [84]. Hence, under conditions of fasting, glucagon contributes to energy homeostasis by elevating circulating levels of glucose and promoting ketogenesis. However, there are important differences between species in this area. First, high doses of glucagon also activate the sympathetic nervous system [85]. Since most studies have investigated the impact of glucagon at doses of 1 mg in humans, resulting in hugely supraphysiological plasma concentrations, many of the published effects of glucagon in humans may be owing to sympathetic nervous system activation [72] and this may also include brown adipose tissue activation. Second, it appears that the GCGR is not expressed in human adipose tissue at all or in the white adipose tissue of mice, while rat adipose tissues seems to have expression this receptor [74]. Rodent studies, therefore, have limited translational value regarding the impact of glucagon on lipolysis.
Central effects of glucagon
The first evidence pointing to a role of the brain in glucose metabolism dates back to the work of Claude Bernard, who in 1854 showed that puncturing the floor of the fourth ventricle increases blood glucose in rodents [86]. Just over 100 years later, it was observed in rabbits that stimulation of the medial hypothalamic area increases blood glucose, while stimulation of the lateral hypothalamus has the opposite effect [87]. More recently, glucose-sensing neurons have been identified in various brain regions, including the hypothalamus, the nucleus tractus solitarius and the area postrema, and are implicated in the regulation of systemic energy metabolism. The molecular events leading to activation or silencing of glucose-sensitive neurons are surprisingly similar to those of the endocrine pancreas and include neuronal uptake of glucose via GLUT2, conversion to pyruvate (and finally ATP) via glucokinase, and modulation of neuronal firing mediated by changes in KATP channel activity, cell depolarisation and Ca2+ influx. While glucose-activated neurons increase their firing rate at high ATP levels via KATP channel-induced membrane depolarisation and increased Ca2+ influx, glucose-inhibited neurons are silent under high ATP levels and are activated when glucose levels (and hence ATP levels) are low [88]. Several studies support the notion that glucagon itself may also influence glucose metabolism via central nervous system (CNS)-associated mechanisms. Glucagon may cross the blood–brain barrier [89] (through circumventricular organs) and, when injected into the third ventricle, inhibits food intake in rodents [90] and birds [91], potentially via mechanisms that include inhibition of protein kinase A (PKA)/Ca2+-calmodulin-dependent protein kinase β (CaMKKβ), AMP-activated protein kinase (AMPK) and agouti-related protein [92]. In line with these observations, glucagon-induced inhibition of food intake is blunted upon inhibition of hypothalamic PKA and AMPK signalling [92]. Beyond the ability of glucagon to decrease food intake via central mechanisms, its administration into the third ventricle increases blood glucose in rodents, birds and dogs, and this is paralleled by a decrease in liver glycogen [93]. Interestingly, the ability of centrally applied glucagon to increase hepatic glycogenolysis persists after vagotomy, but is blocked by pancreatectomy or spinal cord transection [94]. Hence, these data suggest that centrally administered glucagon stimulates pancreatic glucagon secretion to increase glucose levels via the action of pancreatic glucagon on hepatic glucose production and glycogenolysis [94]. The wiring is not established but may involve the sympathetic nervous system. In rats, administration of glucagon into the mediobasal hypothalamus (MBH) improves glucose tolerance by silencing de novo glucose production in the liver [95]. The glucose-lowering effect of MBH-administered glucagon was mediated via GCGR–PKA signalling, depended on functional KATP channels in the MBH and vanished upon hepatic vagotomy [95]. Therefore, it is well established that glucagon has effects on metabolism when administered directly into the CNS, but the physiological relevance of these mechanisms is yet to be established.
Glucagon in disease
Hyperglucagonaemia contributes to a variety of diseases
Hyperglucagonaemia has been reported in various diseases/conditions including type 2 diabetes, type 1 diabetes, obesity, fatty liver diseases and kidney diseases [22]. The last 100 years of research have led to numerous hypotheses and speculations on glucagon’s potential role in these diseases. Increased glucagon may be explained by the following: (1) increased secretion; (2) decreased degradation/extraction/clearance; and (3) inaccurate measurements due to assay cross-reactivity with other glucagon-like molecules (see Fig. 5). Another important factor is in what setting the hyperglucagonaemia is observed, for example under fasting conditions, or postprandially after an OGTT or mixed meal. Here we summarise specific aspects regarding the role of glucagon in specific pathophysiological states.

Hyperglucagonaemia: causes and consequences. Increased glucagon may be explained by: (1) increased secretion; (2) decreased degradation/extraction/clearance; and (3) inaccurate measurements due to assay cross-reactivity with other glucagon-like molecules (not shown). The image depicts some potential causes for diabetogenic hyperglucagonaemia (glucagon depicted as orange hexagons). In the liver, glucagon sensitivity may be impaired in individuals with fatty liver disease (disruption of the liver–alpha cell axis). Glucagon resistance may involve downregulation of amino acid transporters and impairment of glucagon-induced non-transcriptional activation of ureagenesis in hepatocytes. The resulting phenotype is increased plasma levels of amino acids and, perhaps also, lipids, which together drives hypersecretion of glucagon and the ensuing increase in hepatic glucose production. Extraction of glucagon may be carried out by several organs including the kidney, brain and liver, and it may be speculated that, for example, chronic renal failure might contribute to hyperglucagonaemia. Within the islets, alpha cells may react paradoxically to increased glucose levels; following the transport of glucose molecules (depicted as blue circles) across the cell membrane of pancreatic alpha cells, glucagon secretion may be increased rather than suppressed (whether this holds true in humans has not yet been determined). The discrepancies in findings on whether glucose stimulates or inhibits glucagon secretion may be owing to differences in experimental settings (e.g. in vitro vs in vivo) and species (e.g. rats vs humans) [127]. Finally, obesity and diabetes have been associated with reduced GLP-1 secretion and, given that part of the glucose-lowering effect of this hormone is mediated through its glucagonostatic effects, impaired GLP-1 secretion may contribute to hyperglucagonaemia. PLC, phospholipase C. Created with BioRender.com. This figure is available as part of a downloadable slideset
Glucagon in diabetes
Raskin and Unger reported hyperglucagonaemia in individuals with diabetes more than four decades ago [11] and this has been replicated in numerous studies [13, 20, 27]. Hyperglucagonaemia is generally found both during fasting and feeding conditions in individuals with type 2 diabetes [96]. It has also been demonstrated that GCGR antagonists lower glucose and HbA1c levels in both rodent models of type 2 diabetes and individuals with type 2 diabetes, suggesting that hyperglucagonaemia directly contributes to hyperglycaemia in type 2 diabetes [97, 98].
However, postprandial hyperglucagonaemia in type 2 diabetes (in response to OGTT) is not consistently observed. In a cross-sectional trial in which plasma glucagon was measured during an OGTT in >1400 individuals, increased glucagon was observed at fasting and 30 min after the OGTT in individuals with type 2 diabetes compared with those without, but levels at a later time point were similar between the two groups [26]. In agreement with other observations, these data suggest that lack of glucagon suppression may not be a central feature of diabetes [99]. Instead, fasting hyperglucagonaemia appears to be associated with hepatic insulin resistance or, as shown in other studies, hepatic steatosis [83, 100]. Thus, it has been suggested that the hyperglucagonaemia observed with type 2 diabetes may be owing to concurrent liver disease, specifically hepatic steatosis, causing impairment of the liver–alpha cell axis (owing to desensitised hepatic GCGR signalling), resulting in hyperaminoacidaemia and, thereby, increased plasma glucagon both in the fasting and postprandial state [99]. It should be noted that, in the authors’ opinion, a mixed meal as a test stimulus may better reflect the dynamic changes in plasma glucagon that occur in a real-world setting; following a mixed meal, glucagon secretion may increase due to the protein and, potentially, lipid content of the meal. OGTT-induced changes in glucagon secretion, therefore, may not reflect daily living.
Hyperglucagonaemia has also been reported in newly diagnosed individuals with type 1 diabetes and in animal models of type 1 diabetes. In individuals with well-controlled type 1 diabetes, hyperglucagonaemia may not be evident and may only result from inadequate insulin therapy and/or increased neural activity (reflecting a catabolic stress condition). Following on from this, a recent study in individuals with type 1 diabetes treated with a GCGR antagonist demonstrated unimpressive effects of the therapy on blood glucose levels [101].
Glucagon in liver diseases
Individuals with cirrhosis have been shown to have hyperglucagonaemia in some studies, although in others this was not evident [102, 103]. Cirrhosis is somewhat extreme (compared with simple steatosis) since, besides the liver disease, which disrupts the liver–alpha cell axis, individuals with cirrhosis are at high risk of kidney disease, which is often associated with hyperglucagonaemia [104]. Whether there are differences in hepatic extraction of glucagon (e.g. between individuals with and without diabetes) is controversial [105, 106] and the disparity of findings in this area may be related to the choice of glucagon assay [15]. In addition, for individuals with cirrhosis, variation in shunting between the splanchnic and the systemic circulation may also contribute to hepatic glucagon extraction. A third element to if and how glucagon is extracted by the liver may involve GCGR-mediated endocytosis (i.e. hepatic clearance), although this is controversial [107]. Findings of hyperglucagonaemia are more consistently reported in individuals with NAFLD independent of type 2 diabetes [99]. A major confounder is the close association between NAFLD and obesity and, hence, a key question is whether obesity or NAFLD drives hyperglucagonaemia. BMI-matched individuals with and without NAFLD may be studied to answer this question. Preliminary data from the authors’ laboratories suggest that both obesity and NAFLD may contribute to hyperglucagonaemia, although via different mechanism(s) (unpublished results, N. J. Wewer Albrechtsen). A disrupted liver–alpha cell axis has been observed in humans with NAFLD, implying that elevated amino acid levels (and potentially fatty acids) as opposed to dysfunctional glucose sensing by the alpha cells are responsible for hyperglucagonaemia [42].
Glucagon in other diseases
As mentioned above, individuals with chronic kidney disease have hyperglucagonaemia but studies have indicated that this is mainly due to the accumulation of N-terminally extended glucagon 1-61. Glucagon 1-61 a proglucagon product that may have some biological impact through the GCGR [18]. In general, altered clearance of glucagon in various diseases does not appear to be responsible for increased glucagon levels [106]; however, the recently demonstrated importance of activity of the protease neprilysin may have been underestimated [108]. Levels of neprilysin may vary with concurrent metabolic disease; notably, inhibitors of neprilysin are used in heart failure and, in this context, metabolic benefits have been reported.
Glucagon agonism, antagonism or co-agonism
The importance of hyperglucagonaemia for the pathogenesis of type 2 diabetes has already been emphasised, and consistent with this, hepatic glucose production of glucagon is increased in individuals with type 2 diabetes compared with individuals without diabetes of a similar age and with a similar BMI. In addition, somatostatin-induced inhibition of glucagon lowers blood glucose in insulin-deficient dogs and humans with type 1 diabetes [109]. Mice deficient for the GCGR may be protected from diet-induced obesity (DIO) and show improved glucose tolerance and enhanced insulin sensitivity [110]. Interestingly, GCGR-deficient mice are resistant to streptozotocin (STZ)-induced beta cell destruction and hyperglycaemia [111], which led to the suggestion that the presence of glucagon might be more important for the hyperglycaemia associated with type 1 diabetes than the lack of insulin [13]. However, these findings are in contrast to the demonstration that diabetes is an immediate consequence of partial or total pancreatectomy in dogs, rabbits, rodents and humans [112]. Furthermore, deletion of the GCGR in genetically induced insulin-deficient mice cannot rescue hyperglycaemia [113]. Blocking monoclonal antibodies against GCGR also had very limited effect on glucose control in humans with type 1 diabetes [101]. Nevertheless, inhibition of glucagon signalling has consistently been demonstrated to improve glucose control in mice with DIO, ob/ob mice, db/db mice, STZ-induced rat models of diabetes, Zucker rats, rabbits, dogs, monkeys and humans, which supports the therapeutic value of GCGR inhibition for the treatment of type 2 diabetes [13].
In light of glucagon’s ability to increase glucose production, and the documented beneficial effects of glucagon signal inhibition for the treatment of type 2 diabetes, at first glance it seems counter-intuitive that glucagon could be used in unimolecular combinations with GLP-1 to treat obesity and type 2 diabetes [114]. The rationale for the first engineered GLP-1/GCGR co-agonist [114] originated from the observation that chronic administration of a water-soluble form of glucagon not only decreased body weight in mice with DIO, but also improved glucose tolerance with an efficacy similar to that obtained with exendin-4 [70]. Consistent with the non-glycaemic effects of glucagon to inhibit food intake, enhance lipid metabolism and accelerate energy expenditure [70], a single-molecule GLP-1/GCGR co-agonist improved body weight and glucose control in mice with DIO beyond that observed with GLP-1 receptor agonism alone [114, 115]. That GLP-1/GCGR co-agonism can improve glucose, lipid and energy metabolism has subsequently also been demonstrated in human studies [116, 117], and a series of co-agonists have advanced to clinical development for the treatment of type 2 diabetes, non-alcoholic steatohepatitis and obesity (as reviewed previously [117]). Building upon these findings, the addition of glucagon action has also been demonstrated to enhance the metabolic effects of GLP-1/GIP receptor co-agonism [118]. Similar to the dual GLP-1/GCGR agonists, a series of GLP-1/GIP/GCGR tri-agonists have made it to clinical development [117] and have shown favourable efficacy and safety in clinical studies [119, 120].
Future perspective of glucagon
Although glucagon has been studied for more than 100 years, its main physiological actions are still widely discussed and currently under investigation. While glucagon’s role in hepatic glucose metabolism, and the regulation of alpha cell glucagon secretion by glucose have been intensely investigated, numerous other aspects of glucagon biology have received less attention. Key areas including the role of glucagon in the kidney, the effect of glucagon on the heart, and the question of the importance of GCGR signalling for brain-associated metabolism and function including appetite and food intake, remain unresolved. Also, the recent introduction of the concept of glucagon sensitivity [121] will probably result in studies on its importance. Of special clinical interest is the development of therapeutics based on glucagon’s metabolic actions, perhaps in particular in combination with GLP-1 receptor agonists, which may have superior actions, especially in those with the metabolic liver diseases that often accompany obesity and/or type 2 diabetes. In this context, knowledge about the cardiovascular consequences of glucagon agonism is essential [122]. The future use of GCGR agonism will, therefore, probably not be limited to the prevention of hypoglycaemia.

Abbreviations
- AMPK:
-
AMP-activated protein kinase
- CNS:
-
Central nervous system
- DIO:
-
Diet-induced obesity
- FGF21:
-
Fibroblast growth factor 21
- GCGR:
-
Glucagon receptor
- GIP:
-
Glucose-dependent insulinotropic polypeptide
- GLP-1:
-
Glucagon-like peptide 1
- KATP channel:
-
ATP-sensitive potassium channel
- MBH:
-
Mediobasal hypothalamus
- NAFLD:
-
Non-alcoholic fatty liver disease
- PKA:
-
Protein kinase A
- STZ:
-
Streptozotocin
References
Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA (1922) Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J 12(3):141–146
Collip JB (1923) Delayed manifestation of the physiological effects of insulin following the administration of certain pancreatic extracts. Am J Physiol 63:February
Fisher NF (1923) I. Preparation of insulin. Am J Physiol Legacy Content 67(1):57–64. https://doi.org/10.1152/ajplegacy.1923.67.1.57
Kimball CP, Murlin JR (1923) Aqueous extracts of pancreas: III. Some precipitation reactions of insulin. J Biol Chem 58(1):337–346. https://doi.org/10.1016/S0021-9258(18)85474-6
Sutherland E (1950) The effect of the hyperglycemic factor in the pancreas and of epinephrine on glycogenolysis. Recent Prog Horm Res 172(737). https://doi.org/10.1016/B978-0-12-571105-0.50016-2
Staub A, Sinn L, Behrens OK (1953) Purification and crystallization of hyperglycemic glycogenolytic factor (HGF). Science 117(3049):628–629. https://doi.org/10.1126/science.117.3049.628
Bromer WW, Sinn LG, Staub A, Behrens OK (1957) The amino acid sequence of glucagon. Diabetes 6(3):234–238. https://doi.org/10.2337/diab.6.3.234
Sutherland EW, Cori CF, Haynes R, Olsen N et al (1949) Purification of the hyperglycemic-glycogenolytic factor from insulin and from gastric mucosa. J Biol Chem 180(2):825–837. https://doi.org/10.1016/S0021-9258(18)56702-8
Vuylsteke CA, Cornelis G, de Duve C (1952) Influence du traitement au cobalt sur le contenu en facteur H-G du pancreas de Cobaye. Arch Int Physiol 60(1):128–131. https://doi.org/10.3109/13813455209145047. ([article in French])
Unger RH, Eisentraut AM, McCall MS, Keller S, Lanz HC, Madison LL (1959) Glucagon antibodies and their use for immunoassay for glucagon. Proc Soc Exp Biol Med 102:621–623. https://doi.org/10.3181/00379727-102-25338
Raskin P, Unger RH (1978) Hyperglucagonemia and its suppression. N Engl J Med 299(9):433–436. https://doi.org/10.1056/NEJM197808312990901
Unger RH, Aguilar-Parada E, Muller WA, Eisentraut AM (1970) Studies of pancreatic alpha cell function in normal and diabetic subjects. J Clin Invest 49(4):837–848. https://doi.org/10.1172/JCI106297
Unger RH, Cherrington AD (2012) Glucagonocentric restructuring of diabetes: a pathophysiologic and therapeutic makeover. J Clin Invest 122(1):4–12. https://doi.org/10.1172/JCI60016
Sutherland EW, de Duve C (1948) Origin and distribution of the hyperglycemic-glycogenolytic factor of the pancreas. J Biol Chem 175:663–674. https://doi.org/10.1016/S0021-9258(18)57183-0
Holst JJ, WewerAlbrechtsen NJ (2019) Methods and guidelines for measurement of glucagon in plasma. Int J Mol Sci 20(21):5416. https://doi.org/10.3390/ijms20215416
Unger RH (1973) Radioimmunoassay of glucagon. Metab Clin Exp 22(8):979–985. https://doi.org/10.1016/0026-0495(73)90215-1
Mashiter K, Harding PE, Chou M et al (1975) Persistent pancreatic glucagon but not insulin response to arginine in pancreatectomized dogs. Endocrinology 96(3):678–693. https://doi.org/10.1210/endo-96-3-678
Wewer Albrechtsen NJ, Kuhre RE, Hornburg D et al (2017) Circulating glucagon 1–61 regulates blood glucose by increasing insulin secretion and hepatic glucose production. Cell Rep 21(6):1452–1460. https://doi.org/10.1016/j.celrep.2017.10.034
Heding LG (1971) Radioimmunological determination of pancreatic and gut glucagon in plasma. Diabetologia 7(1):10–19. https://doi.org/10.1007/BF02346248
Wewer Albrechtsen NJ, Hartmann B, Veedfald S et al (2014) Hyperglucagonaemia analysed by glucagon sandwich ELISA: nonspecific interference or truly elevated levels? Diabetologia 57(9):1919–1926. https://doi.org/10.1007/s00125-014-3283-z
Alexiadou K, Cuenco J, Howard J et al (2020) Proglucagon peptide secretion profiles in type 2 diabetes before and after bariatric surgery: 1-year prospective study. BMJ Open Diabetes Res Care 8(1):e001076. https://doi.org/10.1136/bmjdrc-2019-001076
Wewer Albrechtsen NJ, Kuhre RE, Pedersen J, Knop FK, Holst JJ (2016) The biology of glucagon and the consequences of hyperglucagonemia. Biomark Med 10(11):1141–1151. https://doi.org/10.2217/bmm-2016-0090
Unger RH, Orci L (1975) The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet 1(7897):14–16. https://doi.org/10.1016/s0140-6736(75)92375-2
Flattem N, Igawa K, Shiota M, Emshwiller MG, Neal DW, Cherrington AD (2001) Alpha- and beta-cell responses to small changes in plasma glucose in the conscious dog. Diabetes 50(2):367–375. https://doi.org/10.2337/diabetes.50.2.367
Doliba NM, Rozo AV, Roman J et al (2022) α Cell dysfunction in islets from nondiabetic, glutamic acid decarboxylase autoantibody-positive individuals. J Clin Invest 132(11):e156243. https://doi.org/10.1172/jci156243
Færch K, Vistisen D, Pacini G et al (2016) Insulin resistance is accompanied by increased fasting glucagon and delayed glucagon suppression in individuals with normal and impaired glucose Regulation. Diabetes 65(11):3473–3481. https://doi.org/10.2337/db16-0240
Knop FK, Vilsbøll T, Madsbad S, Holst JJ, Krarup T (2007) Inappropriate suppression of glucagon during OGTT but not during isoglycaemic i.v. glucose infusion contributes to the reduced incretin effect in type 2 diabetes mellitus. Diabetologia 50(4):797–805. https://doi.org/10.1007/s00125-006-0566-z
Markova M, Hornemann S, Sucher S et al (2018) Rate of appearance of amino acids after a meal regulates insulin and glucagon secretion in patients with type 2 diabetes: a randomized clinical trial. Am J Clin Nutr 108(2):279–291. https://doi.org/10.1093/ajcn/nqy100
Wewer Albrechtsen NJ, Pedersen J, Galsgaard KD et al (2019) The liver-alpha-cell axis and type 2 diabetes. Endocr Rev 40(5):1353–1366. https://doi.org/10.1210/er.2018-00251
Christensen M, Vedtofte L, Holst JJ, Vilsboll T, Knop FK (2011) Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes 60(12):3103–3109. https://doi.org/10.2337/db11-0979
Chae H, Augustin R, Gatineau E et al (2020) SGLT2 is not expressed in pancreatic alpha- and beta-cells, and its inhibition does not directly affect glucagon and insulin secretion in rodents and humans. Mol Metab 42:101071. https://doi.org/10.1016/j.molmet.2020.101071
Rorsman P, Ramracheya R, Rorsman NJ, Zhang Q (2014) ATP-regulated potassium channels and voltage-gated calcium channels in pancreatic alpha and beta cells: similar functions but reciprocal effects on secretion. Diabetologia 57(9):1749–1761. https://doi.org/10.1007/s00125-014-3279-8
Dai C, Walker JT, Shostak A et al (2020) Dapagliflozin does not directly affect human α or β cells. Endocrinology 161(8):bqaa080. https://doi.org/10.1210/endocr/bqaa080
Gylfe E, Gilon P (2014) Glucose regulation of glucagon secretion. Diabetes Res Clin Pract 103(1):1–10. https://doi.org/10.1016/j.diabres.2013.11.019
Rorsman P, Huising MO (2018) The somatostatin-secreting pancreatic delta-cell in health and disease. Nat Rev Endocrinol 14(7):404–414. https://doi.org/10.1038/s41574-018-0020-6
Maruszczak K, Rasmussen C, Ceutz FR et al (2022) Arginine-induced glucagon secretion and glucagon-induced enhancement of amino acid catabolism are not influenced by ambient glucose levels in mice. Am J Physiol Endocrinol Metab 323(3):E207–E214. https://doi.org/10.1152/ajpendo.00122.2022
Rocha DM, Faloona GR, Unger RH (1972) Glucagon-stimulating activity of 20 amino acids in dogs. J Clin Invest 51(9):2346–2351. https://doi.org/10.1172/JCI107046
Briant LJB, Dodd MS, Chibalina MV et al (2018) CPT1a-dependent long-chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Rep 23(11):3300–3311. https://doi.org/10.1016/j.celrep.2018.05.035
Dean ED (2020) A primary role for α-cells as amino acid sensors. Diabetes 69(4):542–549. https://doi.org/10.2337/dbi19-0021
Muller WA, Faloona GR, Unger RH (1971) The effect of alanine on glucagon secretion. J Clin Invest 50(10):2215–2218. https://doi.org/10.1172/JCI106716
Galsgaard KD, Winther-Sorensen M, Pedersen J et al (2019) Glucose and amino acid metabolism in mice depend mutually on glucagon and insulin receptor signaling. Am J Physiol Endocrinol Metab 316(4):E660–E673. https://doi.org/10.1152/ajpendo.00410.2018
Richter MM, Galsgaard KD, Elmelund E et al (2022) The liver-alpha-cell axis in health and in disease. Diabetes 71(9):1852–1861. https://doi.org/10.2337/dbi22-0004
Suppli MP, Bagger JI, Lund A et al (2020) Glucagon resistance at the level of amino acid turnover in obese subjects with hepatic steatosis. Diabetes 69(6):1090–1099. https://doi.org/10.2337/db19-0715
Campbell JE, Newgard CB (2021) Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol 22(2):142–158. https://doi.org/10.1038/s41580-020-00317-7
Svendsen B, Larsen O, Gabe MBN et al (2018) Insulin secretion depends on intra-islet glucagon signaling. Cell Rep 25(5):1127–1134. https://doi.org/10.1016/j.celrep.2018.10.018. (e1122)
Strowski MZ, Parmar RM, Blake AD, Schaeffer JM (2000) Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology 141(1):111–117. https://doi.org/10.1210/endo.141.1.7263
Rorsman P, Braun M, Zhang Q (2012) Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell calcium 51(3–4):300–308. https://doi.org/10.1016/j.ceca.2011.11.006
Gilon P (2020) The role of alpha-cells in islet function and glucose homeostasis in health and type 2 diabetes. J Mol Biol 432(5):1367–1394. https://doi.org/10.1016/j.jmb.2020.01.004
Xu SFS, Andersen DB, Izarzugaza JMG, Kuhre RE, Holst JJ (2020) In the rat pancreas, somatostatin tonically inhibits glucagon secretion and is required for glucose-induced inhibition of glucagon secretion. Acta Physiol 229(3):e13464. https://doi.org/10.1111/apha.13464
Vergari E, Knudsen JG, Ramracheya R et al (2019) Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat Commun 10(1):139. https://doi.org/10.1038/s41467-018-08193-8
Andersen DB, Holst JJ (2022) Peptides in the regulation of glucagon secretion. Peptides 148:170683. https://doi.org/10.1016/j.peptides.2021.170683
Adriaenssens AE, Svendsen B, Lam BY et al (2016) Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59(10):2156–2165. https://doi.org/10.1007/s00125-016-4033-1
Holst JJ, Schwartz TW, Knuhtsen S, Jensen SL, Nielsen OV (1986) Autonomic nervous control of the endocrine secretion from the isolated, perfused pig pancreas. J Auton Nerv Syst 17(1):71–84. https://doi.org/10.1016/0165-1838(86)90045-7
Holst JJ (1990) Peptidergic mechanisms in the pancreas. Arch Int Pharmacodyn Ther 303(252–69):252–269
Harp JB, Yancopoulos GD, Gromada J (2016) Glucagon orchestrates stress-induced hyperglycaemia. Diabetes Obes Metab 18(7):648–653. https://doi.org/10.1111/dom.12668
Galbo H, Richter EA, Christensen NJ, Holst JJ (1978) Sympathetic control of metabolic and hormonal responses to exercise in rats. Acta Physiol Scand 102(4):441–449. https://doi.org/10.1111/j.1748-1716.1978.tb06092.x
Palmer JP, Ensinck JW (1975) Stimulation of glucagon secretion by ethanol-induced hypoglycemia in man. Diabetes 24(3):295–300. https://doi.org/10.2337/diab.24.3.295
Mumme L, Breuer TGK, Rohrer S et al (2017) Defects in alpha-cell function in patients with diabetes due to chronic pancreatitis compared with patients with type 2 diabetes and healthy individuals. Diabetes Care 40(10):1314–1322. https://doi.org/10.2337/dc17-0792
Meyer C, Grossmann R, Mitrakou A et al (1998) Effects of autonomic neuropathy on counterregulation and awareness of hypoglycemia in type 1 diabetic patients. Diabetes Care 21(11):1960–1966. https://doi.org/10.2337/diacare.21.11.1960
Biggers DW, Myers SR, Neal D et al (1989) Role of brain in counterregulation of insulin-induced hypoglycemia in dogs. Diabetes 38(1):7–16. https://doi.org/10.2337/diab.38.1.7
Lund A, Vilsboll T, Bagger JI, Holst JJ, Knop FK (2011) The separate and combined impact of the intestinal hormones, GIP, GLP-1 and GLP-2, on glucagon secretion in type 2 diabetes. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00665.2010
Hare KJ, Vilsboll T, Asmar M, Deacon CF, Knop FK, Holst JJ (2010) The glucagonostatic and insulinotropic effects of glucagon-like peptide-1 contribute equally to its glucose-lowering action. Diabetes 59(7):1765–1770. https://doi.org/10.2337/db09-1414
Holst JJ, Pedersen J, Wewer Albrechtsen NJ, Knop FK (2017) The gut: a key to the pathogenesis of type 2 diabetes? Metab Syndr Relat Disord 15(6):259–262. https://doi.org/10.1089/met.2017.0015
Nauck MA, Heimesaat MM, Behle K et al (2002) Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab 87(3):1239–1246. https://doi.org/10.1210/jcem.87.3.8355
Ding WG, Renstrom E, Rorsman P, Buschard K, Gromada J (1997) Glucagon-like peptide I and glucose-dependent insulinotropic polypeptide stimulate Ca2+-induced secretion in rat alpha-cells by a protein kinase A-mediated mechanism. Diabetes 46(5):792–800. https://doi.org/10.2337/diab.46.5.792
Orgaard A, Holst JJ (2017) The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia 60(9):1731–1739. https://doi.org/10.1007/s00125-017-4315-2
Holst JJ (2007) The physiology of glucagon-like peptide 1. Physiol Rev 87(4):1409–1439. https://doi.org/10.1152/physrev.00034.2006
Vasileva A, Marx T, Beaudry JL, Stern JH (2022) Glucagon receptor signaling at white adipose tissue does not regulate lipolysis. Am J Physiol Endocrinol Metab 323(4):E389–E401. https://doi.org/10.1152/ajpendo.00078.2022
Habegger KM, Stemmer K, Cheng C et al (2013) Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes 62(5):1453–1463. https://doi.org/10.2337/db12-1116
Muller TD, Finan B, Clemmensen C, DiMarchi RD, Tschop MH (2017) The new biology and pharmacology of glucagon. Physiol Rev 97(2):721–766. https://doi.org/10.1152/physrev.00025.2016
Capozzi ME, Wait JB, Koech J et al (2019) Glucagon lowers glycemia when β cells are active. JCI Insight 4(16):e129954. https://doi.org/10.1172/jci.insight.129954
Takayama S, Nakajima Y, Toma S, Sakamoto T (1995) Increased muscle sympathetic nerve activity after glucagon administration in man. J Auton Nervous Syst 54(2):171–175. https://doi.org/10.1016/0165-1838(95)00007-K
Edgerton DS, Kraft G, Smith M, Farmer B, Williams PE, Cherrington AD (2023) A physiologic increase in brain glucagon action alters the hepatic gluconeogenic glycogenolytic ratio but not glucagon’s overall effect on glucose production. Am J Physiol Endocrinol Metab 324(2):E199–E208. https://doi.org/10.1152/ajpendo.00304.2022
Bomholt AB, Johansen CD, Christensen JB et al (2022) Evaluation of commercially available glucagon receptor antibodies and glucagon receptor expression. Commun Biol 5(1):1278. https://doi.org/10.1038/s42003-022-04242-7
Vilstrup H, Hansen BA, Almdal TP (1990) Glucagon increases hepatic efficacy for urea synthesis. J Hepatol 10(1):46–50. https://doi.org/10.1016/0168-8278(90)90072-Y
Elmelund E, Galsgaard KD, Johansen CD et al (2022) Opposing effects of chronic glucagon receptor agonism and antagonism on amino acids, hepatic gene expression, and alpha cells. iScience 25(11):105296. https://doi.org/10.1016/j.isci.2022.105296
Kraft G, Coate KC, Winnick JJ et al (2017) Glucagon’s effect on liver protein metabolism in vivo. Am J Physiol Endocrinol Metab 313(3):E263-e272. https://doi.org/10.1152/ajpendo.00045.2017
Solloway Mark J, Madjidi A, Gu C et al (2015) Glucagon couples hepatic amino acid catabolism to mTOR-dependent regulation of α-cell mass. Cell Rep 12(3):495–510. https://doi.org/10.1016/j.celrep.2015.06.034
Galsgaard KD, Pedersen J, Knop FK, Holst JJ, WewerAlbrechtsen NJ (2019) Glucagon receptor signaling and lipid metabolism. Front Physiol 10:413. https://doi.org/10.3389/fphys.2019.00413
Caren R, Carbo L (1956) Pancreatic alpha-cell function in relation to cholesterol metabolism. J Clin Endocrinol Metab 16(4):507–516. https://doi.org/10.1210/jcem-16-4-507
Brown NF, Salter AM, Fears R, Brindley DN (1989) Glucagon, cyclic AMP and adrenaline stimulate the degradation of low-density lipoprotein by cultured rat hepatocytes. Biochem J 262(2):425–429. https://doi.org/10.1042/bj2620425
Galsgaard KD, Elmelund E, Johansen CD et al (2022) Glucagon receptor antagonism impairs and glucagon receptor agonism enhances triglycerides metabolism in mice. Mol Metab 66:101639. https://doi.org/10.1016/j.molmet.2022.101639
Heebøll S, Risikesan J, Ringgaard S et al (2022) Impaired glucagon-mediated suppression of VLDL-triglyceride secretion in individuals with metabolic dysfunction-associated fatty liver disease (MAFLD). Diabetes 71(11):2402–2411. https://doi.org/10.2337/db22-0313
Gerich JE, Lorenzi M, Bier DM et al (1975) Prevention of human diabetic ketoacidosis by somatostatin. Evidence for an essential role of glucagon. N Engl J Med 292(19):985–989. https://doi.org/10.1056/NEJM197505082921901
Krzeski R, Czyzyk-Krzeska MF, Trzebski A, Millhorn DE (1989) Centrally administered glucagon stimulates sympathetic nerve activity in rat. Brain Res 504(2):297–300. https://doi.org/10.1016/0006-8993(89)91372-3
Bernard C (1854) Leçons de physiologie experimentale appliqués á lá medecine. J-B Baillière, Paris [in French]
Shimazu T, Fukuda A, Ban T (1966) Reciprocal influences of the ventromedial and lateral hypothalamic nuclei on blood glucose level and liver glycogen content. Nature 210(5041):1178–1179. https://doi.org/10.1038/2101178a0
Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE (2002) Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes 51(7):2056–2065. https://doi.org/10.2337/diabetes.51.7.2056
Banks WA, Kastin AJ (1985) Peptides and the blood-brain barrier: lipophilicity as a predictor of permeability. Brain Res Bull 15(3):287–292. https://doi.org/10.1016/0361-9230(85)90153-4
Inokuchi A, Oomura Y, Nishimura H (1984) Effect of intracerebroventricularly infused glucagon on feeding behavior. Physiol Behav 33(3):397–400. https://doi.org/10.1016/0031-9384(84)90160-4
Honda K, Kamisoyama H, Saito N, Kurose Y, Sugahara K, Hasegawa S (2007) Central administration of glucagon suppresses food intake in chicks. Neurosci Lett 416(2):198–201. https://doi.org/10.1016/j.neulet.2007.02.011
Quinones M, Al-Massadi O, Gallego R et al (2015) Hypothalamic CaMKKbeta mediates glucagon anorectic effect and its diet-induced resistance. Mol Metab 4(12):961–970. https://doi.org/10.1016/j.molmet.2015.09.014
Abraham MA, Lam TKT (2016) Glucagon action in the brain. Diabetologia 59(7):1367–1371. https://doi.org/10.1007/s00125-016-3950-3
Agarwala GC, Mishra R, Jaiswal G, Bapat V (1989) Effect of centrally administered glucagon on liver glycogen & enzymes in anaesthetised dogs. Indian J Med Res 90:372–378
Mighiu PI, Yue JT, Filippi BM et al (2013) Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat Med 19(6):766–772. https://doi.org/10.1038/nm.3115
Muller WA, Faloona GR, Aguilar-Parada E, Unger RH (1970) Abnormal alpha-cell function in diabetes. Response to carbohydrate and protein ingestion. N Engl J Med 283(3):109–115. https://doi.org/10.1056/nejm197007162830301
Kazda CM, Garhyan P, Kelly RP et al (2015) A randomized, double-blind, placebo-controlled phase 2 study of the glucagon receptor antagonist LY2409021 in patients with type 2 diabetes. Diabetes Care 37(7):1241–1249. https://doi.org/10.2337/dc15-1643
Johnson DG, Goebel CU, Hruby VJ, Bregman MD, Trivedi D (1982) Hyperglycemia of diabetic rats decreased by a glucagon receptor antagonist. Science 215(4536):1115–1116. https://doi.org/10.1126/science.6278587
Wewer Albrechtsen NJ, Junker AE, Christensen M et al (2017) Hyperglucagonemia correlates with plasma levels of non-branched chained amino acids in patients with liver disease independent of type 2 diabetes. Am J Physiol Gastrointest Liver Physiol 1(314):G91–G96. https://doi.org/10.1152/ajpgi.00216.2017
Wewer Albrechtsen NJ, Færch K, Jensen TM et al (2018) Evidence of a liver-alpha cell axis in humans: hepatic insulin resistance attenuates relationship between fasting plasma glucagon and glucagonotropic amino acids. Diabetologia 61(3):671–680. https://doi.org/10.1007/s00125-017-4535-5
Pettus J, Boeder SC, Christiansen MP et al (2022) Glucagon receptor antagonist volagidemab in type 1 diabetes: a 12-week, randomized, double-blind, phase 2 trial. Nat Med 28(10):2092–2099. https://doi.org/10.1038/s41591-022-02011-x
Sherwin RS, Fisher M, Bessoff J et al (1978) Hyperglucagonemia in cirrhosis: altered secretion and sensitivity to glucagon. Gastroenterology 74(6):1224–1228. https://doi.org/10.1016/0016-5085(78)90696-0
Junker AE, Gluud L, Holst JJ, Knop FK, Vilsboll T (2016) Diabetic and nondiabetic patients with nonalcoholic fatty liver disease have an impaired incretin effect and fasting hyperglucagonaemia. J Intern Med 279(5):485–493. https://doi.org/10.1111/joim.12462
Bilbrey GL, Faloona GR, White MG, Knochel JP (1974) Hyperglucagonemia of renal failure. J Clin Investig 53(3):841–847. https://doi.org/10.1172/jci107624
Röjdmark S, Bloom G, Chou MC, Jaspan JB, Field JB (1978) Hepatic insulin and glucagon extraction after their augmented secretion in dogs. Am J Physiol Endocrinol Metab 235(1):E88. https://doi.org/10.1152/ajpendo.1978.235.1.E88
Grøndahl MFG, Lund AB, Bagger JI et al (2021) Glucagon clearance is preserved in type 2 diabetes. Diabetes 71(1):73–82. https://doi.org/10.2337/db21-0024
Sekar R, Motzler K, Kwon Y et al (2022) Vps37a regulates hepatic glucose production by controlling glucagon receptor localization to endosomes. Cell Metab 34(11):1824–1842. https://doi.org/10.1016/j.cmet.2022.09.022. (e1829)
Kjeldsen SAS, Hansen LH, Esser N et al (2021) Neprilysin inhibition increases glucagon levels in humans and mice with potential effects on amino acid metabolism. J Endocr Soc 5(9):bvab084. https://doi.org/10.1210/jendso/bvab084
Gerich JE, Lorenzi M, Schneider V et al (1974) Effects of somatostatin on plasma glucose and glucagon levels in human diabetes mellitus. Pathophysiologic and therapeutic implications. N Engl J Med 291(11):544–547. https://doi.org/10.1056/NEJM197409122911102
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic α cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci USA 100(3):1438–1443. https://doi.org/10.1073/pnas.0237106100
Conarello SL, Jiang G, Mu J et al (2007) Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia 50(1):142–150. https://doi.org/10.1007/s00125-006-0481-3
Mezza T, Cefalo CMA, Cinti F et al (2020) Endocrine and metabolic insights from pancreatic surgery. Trends Endocrinol Metab 31(10):760–772. https://doi.org/10.1016/j.tem.2020.07.003
Neumann UH, Ho JS, Mojibian M, Covey SD, Charron MJ, Kieffer TJ (2016) Glucagon receptor gene deletion in insulin knockout mice modestly reduces blood glucose and ketones but does not promote survival. Mol Metab 5(8):731–736. https://doi.org/10.1016/j.molmet.2016.05.014
Day JW, Ottaway N, Patterson JT et al (2009) A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol 5(10):749–757. https://doi.org/10.1038/nchembio.209
Henderson SJ, Konkar A, Hornigold DC et al (2016) Robust anti-obesity and metabolic effects of a dual GLP-1/glucagon receptor peptide agonist in rodents and non-human primates. Diabetes Obes Metab 18(12):1176–1190. https://doi.org/10.1111/dom.12735
Tillner J, Posch MG, Wagner F et al (2019) A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes Metab 21(1):120–128. https://doi.org/10.1111/dom.13494
Muller TD, Bluher M, Tschop MH, DiMarchi RD (2022) Anti-obesity drug discovery: advances and challenges. Nat Rev Drug Discov 21(3):201–223. https://doi.org/10.1038/s41573-021-00337-8
Finan B, Yang B, Ottaway N et al (2015) A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med 21(1):27–36. https://doi.org/10.1038/nm.3761
Coskun T, Urva S, Roell WC et al (2022) LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: from discovery to clinical proof of concept. Cell Metab 34(9):1234–1247. https://doi.org/10.1016/j.cmet.2022.07.013. (e1239)
Urva S, Coskun T, Loh MT et al (2022) LY3437943, a novel triple GIP, GLP-1, and glucagon receptor agonist in people with type 2 diabetes: a phase 1b, multicentre, double-blind, placebo-controlled, randomised, multiple-ascending dose trial. Lancet 400(10366):1869–1881. https://doi.org/10.1016/S0140-6736(22)02033-5
Kjeldsen SAS, Richter MM, Jensen NJ et al (2023) Development of a glucagon sensitivity test in humans: pilot data and the GLUSENTIC study protocol. Peptides 161:170938. https://doi.org/10.1016/j.peptides.2022.170938
Ali S, Ussher JR, Baggio LL et al (2015) Cardiomyocyte glucagon receptor signaling modulates outcomes in mice with experimental myocardial infarction. Mol Metab 4(2):132–143. https://doi.org/10.1016/j.molmet.2014.11.005
Petersen KM, Bøgevig S, Holst JJ, Knop FK, Christensen MB (2018) Hemodynamic effects of glucagon: a literature review. J Clin Endocrinol Metab 103(5):1804–1812. https://doi.org/10.1210/jc.2018-00050
Bankir L, Bouby N, Blondeau B, Crambert G (2016) Glucagon actions on the kidney revisited: possible role in potassium homeostasis. Am J Physiol Renal Physiol 311(2):F469–F486. https://doi.org/10.1152/ajprenal.00560.2015
Watanabe O, Atobe Y, Akagi M, Nishi K (1982) Effects of glucagon on myoelectrical activity of the stomach of conscious and anesthetized dogs. Eur J Pharmacol 79(1–2):31–41. https://doi.org/10.1016/0014-2999(82)90572-6
Mochiki E, Suzuki H, Takenoshita S et al (1998) Mechanism of inhibitory effect of glucagon on gastrointestinal motility and cause of side effects of glucagon. J Gastroenterol 33(6):835–841. https://doi.org/10.1007/s005350050184
MacDonald PE, Rorsman P (2023) Metabolic messengers: glucagon. Nat Metab 5(2):186–192. https://doi.org/10.1038/s42255-022-00725-3
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Acknowledgements
We acknowledge the many contributors to the glucagon field, including members from our own groups, without whom this review would not have been made possible. This review was based on the international glucagon conference, entitled ‘A hundred years with glucagon and a hundred more’, held in Copenhagen, Denmark, in November 2022. Figures were created with BioRender.com.
Funding
The review is supported by an unrestricted grant from the Novo Nordic Foundation (NNF21OC0072718) to NJWA in relation to hosting the international glucagon conference in Copenhagen, 1–2 November 2022, in the celebration of glucagon’s 100 year birthday. Furthermore, work in the authors’ laboratories is supported by several funding agencies: NJWA is supported by a grant from European Foundation for the Study of Diabetes Future Leader Award (NNF21SA0072746) and a grant from Independent Research Fund Denmark, Sapere Aude (1052–00003B); EDD is supported by the National Institute of Health (R01DK132669 and R01DK117147); and TDM received funding from the German Research Foundation (DFG TRR296, TRR152, SFB1123 and GRK 2816/1), the German Center for Diabetes Research (DZD e.V.) and the European Research Council ERC-CoG Trusted no. 101044445.
Authors’ relationships and activities
NJWA has received funding from, and served on scientific advisory panels and/or speakers’ bureaus for Boehringer Ingelheim, MSD/MERCK, Novo Nordisk and Mercodia. JJH has served on scientific advisory panels and received speaker fees for Novo Nordisk. ADC’s external relationships include consulting, research advisory boards and research contract: Abvance; Adipo-Pharma; Cellular Longevity, Inc. dba Loyal; Diakard/Diabetica; Fractyl Laboratories, Inc; Novo Nordisk, Inc; Sekki Bio; Senda Biosciences, Inc; Sensulin Labs, LLC; Thetis Pharmaceuticals, LLC; vTv Therapeutics. BF is employed at Novo Nordisk. LLG has received funding, consultant fees from and/or served on scientific advisory panels for Novo Nordisk, Gilead, Becton Dickinson, Sobi, Pfizer and AstraZeneca. JEC receives research funding from Novo Nordisk and Eli Lilly. SRB and TM-MT are shareholders in Zihipp, Ltd. FKK has served on scientific advisory panels and/or been part of speaker’s bureaus for, served as a consultant to and/or received research support from Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Carmot Therapeutics, Eli Lilly, Gubra, Lupin, MedImmune, MSD/Merck, Mundipharma, Norgine, Novo Nordisk, Pharmacosmos, Sanofi, ShouTi, Zealand Pharma and Zucara; and is a minority shareholder in Antag Therapeutics and co-owner of the weight loss clinic Medicinsk Vægttabsbehandling ApS. TDM has received funding from Novo Nordisk. The remaining author (DD) declares that there are no relationships or activities that might bias, or be perceived to bias, their work.
Contribution statement
NJWA, JJH and TDM drafted the first version of the review. ADC, BF, LLG, EDD, JEC, SRB, TM-MT and FKK revised the review for important intellectual content. NJWA drafted figures. All authors approved the version to be published.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wewer Albrechtsen, N.J., Holst, J.J., Cherrington, A.D. et al. 100 years of glucagon and 100 more. Diabetologia 66, 1378–1394 (2023). https://doi.org/10.1007/s00125-023-05947-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-023-05947-y