
mRNA in the Context of Protein Replacement Therapy
 , , , , , and
, , , , , and
Abstract
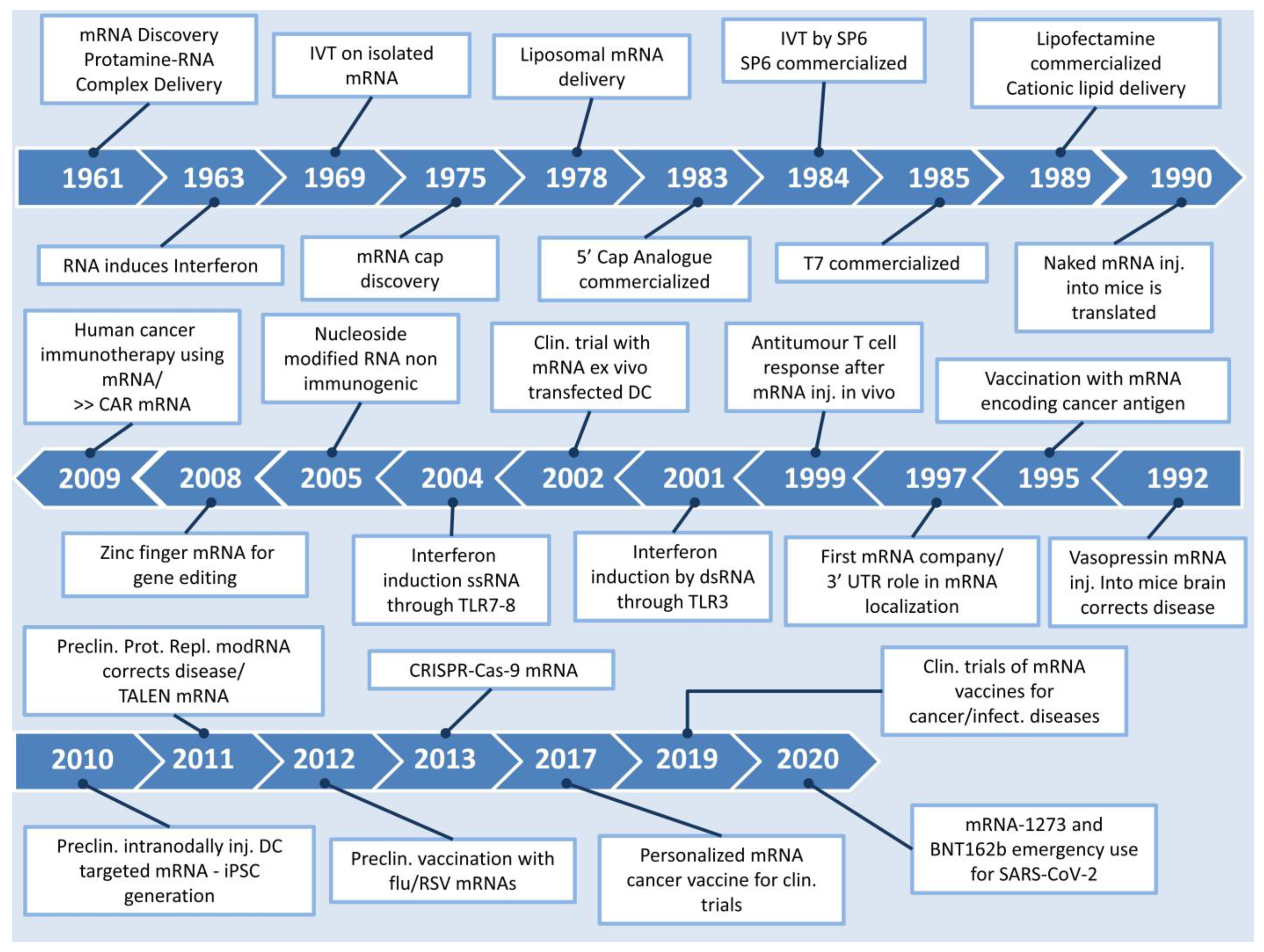
:1. Introduction
Protein Replacement Options—Why Choose mRNA?
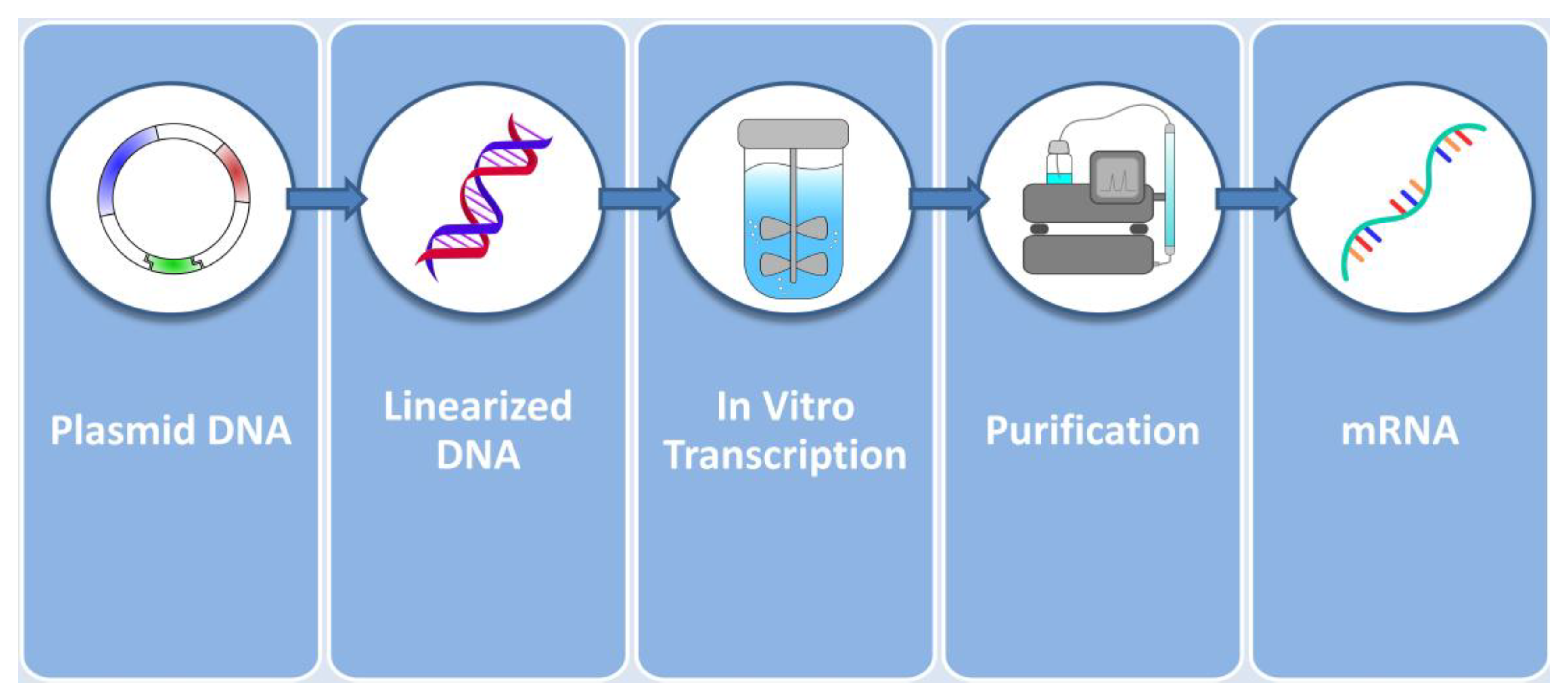
2. Considerations on mRNA Production for Protein Replacement Therapies
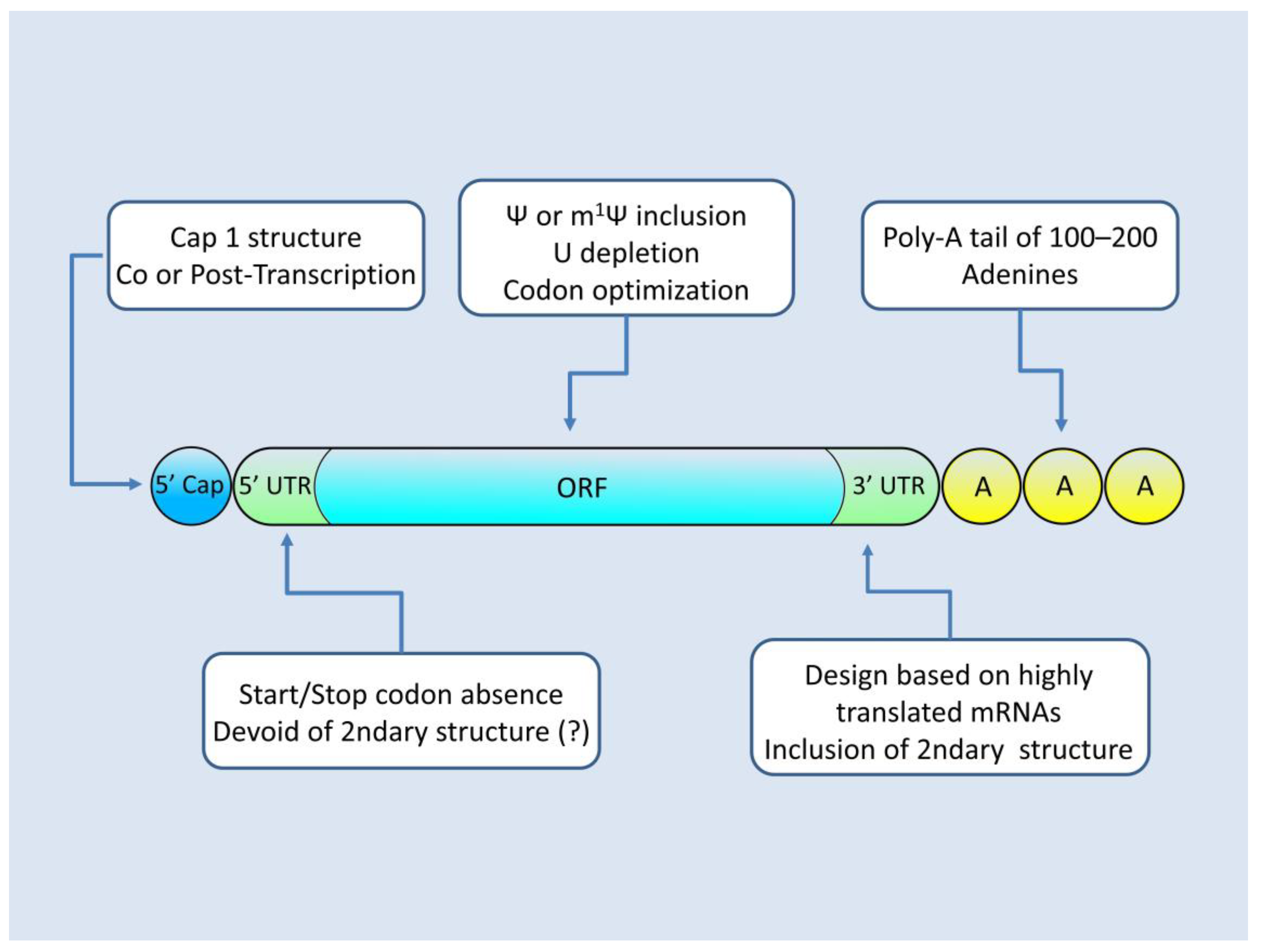
mRNA Architecture for Successful Protein Production
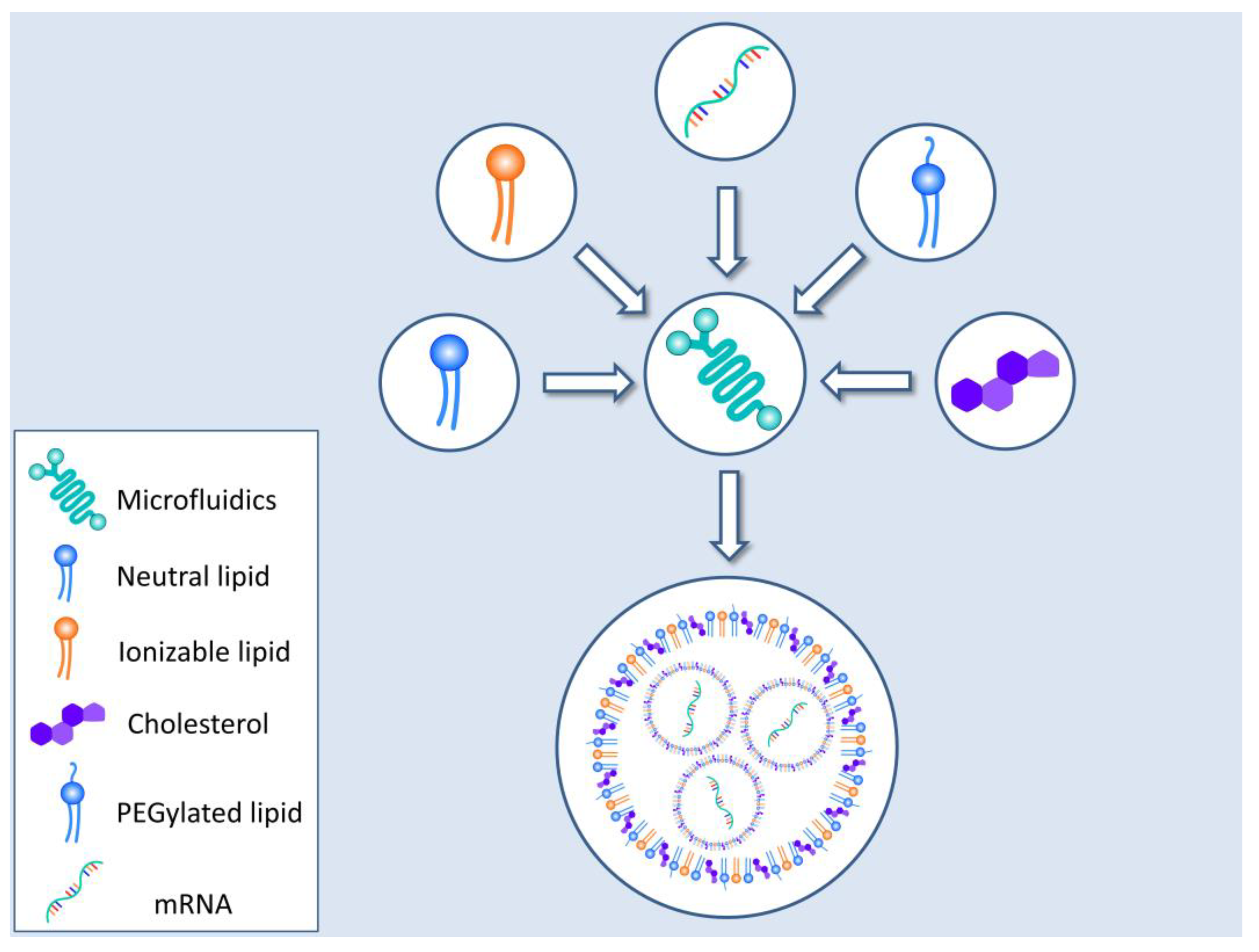
3. Vehicles for Delivering mRNA to the Cells
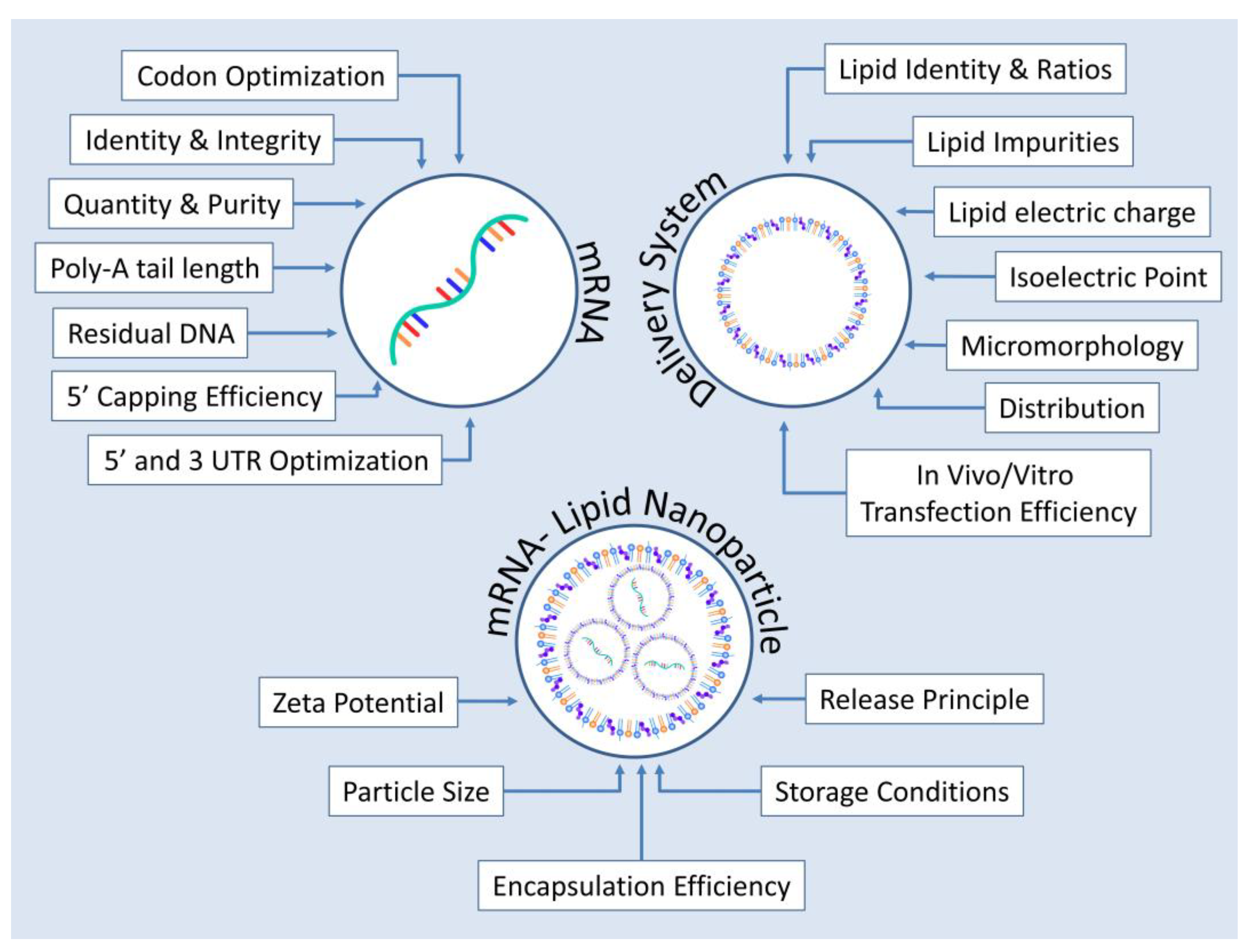
4. Critical Quality Attributes for mRNA-Based Protein Replacement Therapies
5. mRNA-Based Protein Replacement Therapies in Preclinical and Clinical Stage
6. Conclusions and Future Applications
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lim, K.R.Q.; Maruyama, R.; Yokota, T. Eteplirsen in the Treatment of Duchenne Muscular Dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, V.; Wang, A.K. Inotersen: New Promise for the Treatment of Hereditary Transthyretin Amyloidosis. Drug Des. Devel. Ther. 2019, 13, 1515–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, J.; Duggan, S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. [Google Scholar] [CrossRef]
- Mahajan, R. Onasemnogene Abeparvovec for Spinal Muscular Atrophy: The Costlier Drug Ever. Int. J. Appl. Basic Med. Res. 2019, 9, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi Therapeutic for the Treatment of Hereditary Transthyretin-Mediated Amyloidosis. Neurodegener. Dis. Manag. 2019, 9, 5–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majeed, C.N.; Ma, C.D.; Xiao, T.; Rudnick, S.; Bonkovsky, H.L. Spotlight on Givosiran as a Treatment Option for Adults with Acute Hepatic Porphyria: Design, Development, and Place in Therapy. Drug Des. Devel. Ther. 2022, 16, 1827–1845. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct Gene Transfer into Mouse Muscle in Vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of Diabetes Insipidus in Brattleboro Rats: Intrahypothalamic Injection of Vasopressin MRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a Messenger RNA Polynucleotide Vaccine Vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Gorzelany, J.A.; de Souza, M.P. Protein Replacement Therapies for Rare Diseases: A Breeze for Regulatory Approval? Sci. Transl. Med. 2013, 5, 178fs10. [Google Scholar] [CrossRef] [PubMed]
- Baptista, B.; Carapito, R.; Laroui, N.; Pichon, C.; Sousa, F. MRNA, a Revolution in Biomedicine. Pharmaceutics 2021, 13, 2090. [Google Scholar] [CrossRef]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. MRNA-Based Therapeutics: Powerful and Versatile Tools to Combat Diseases. Sig. Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Magadum, A.; Kaur, K.; Zangi, L. MRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into MRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Zangi, L.; Lui, K.O.; von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Später, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R.; et al. Modified MRNA Directs the Fate of Heart Progenitor Cells and Induces Vascular Regeneration after Myocardial Infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N(1)-Methylpseudouridine-Incorporated MRNA Outperforms Pseudouridine-Incorporated MRNA by Providing Enhanced Protein Expression and Reduced Immunogenicity in Mammalian Cell Lines and Mice. J. Control Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Sultana, N.; Magadum, A.; Hadas, Y.; Kondrat, J.; Singh, N.; Youssef, E.; Calderon, D.; Chepurko, E.; Dubois, N.; Hajjar, R.J.; et al. Optimizing Cardiac Delivery of Modified MRNA. Mol. Ther. 2017, 25, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Brenner, S.; Jacob, F.; Meselson, M. An Unstable Intermediate Carrying Information from Genes to Ribosomes for Protein Synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef]
- Smull, C.E.; Mallette, M.F.; Ludwig, E.H. The Use of Basic Proteins to Increase the Infectivity of Enterovirus Ribonucleic Acid. Biochem. Biophys. Res. Commun. 1961, 5, 247–249. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Lane, C.D.; Woodland, H.R.; Marbaix, G. Use of Frog Eggs and Oocytes for the Study of Messenger RNA and Its Translation in Living Cells. Nature 1971, 233, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Muthukrishnan, S.; Both, G.W.; Furuichi, Y.; Shatkin, A.J. 5′-Terminal 7-Methylguanosine in Eukaryotic MRNA Is Required for Translation. Nature 1975, 255, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Dimitriadis, G.J. Translation of Rabbit Globin MRNA Introduced by Liposomes into Mouse Lymphocytes. Nature 1978, 274, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic Liposome-Mediated RNA Transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, L.E.; Malter, J.S. Regulation of Eukaryotic Messenger RNA Turnover. Prog. Nucleic Acid Res. Mol. Biol. 1997, 56, 257–286. [Google Scholar] [CrossRef]
- Zhou, W.Z.; Hoon, D.S.; Huang, S.K.; Fujii, S.; Hashimoto, K.; Morishita, R.; Kaneda, Y. RNA Melanoma Vaccine: Induction of Antitumor Immunity by Human Glycoprotein 100 MRNA Immunization. Hum. Gene Ther. 1999, 10, 2719–2724. [Google Scholar] [CrossRef]
- Heiser, A.; Coleman, D.; Dannull, J.; Yancey, D.; Maurice, M.A.; Lallas, C.D.; Dahm, P.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Autologous Dendritic Cells Transfected with Prostate-Specific Antigen RNA Stimulate CTL Responses against Metastatic Prostate Tumors. J. Clin. Investig. 2002, 109, 409–417. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.-G.; Garbe, C. Direct Injection of Protamine-Protected MRNA: Results of a Phase 1/2 Vaccination Trial in Metastatic Melanoma Patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, O.; Sahin, U. Intranodal Vaccination with Naked Antigen-Encoding RNA Elicits Potent Prophylactic and Therapeutic Antitumoral Immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [Green Version]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.-J.; Stitz, L.; et al. Protective Efficacy of in Vitro Synthesized, Specific MRNA Vaccines against Influenza A Virus Infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral Delivery of Self-Amplifying RNA Vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient Genome Editing in Zebrafish Using a CRISPR-Cas System. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [Green Version]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and Immunogenicity of a MRNA Rabies Vaccine in Healthy Adults: An Open-Label, Non-Randomised, Prospective, First-in-Human Phase 1 Clinical Trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the MRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.B.; Kanevsky, I.; Che, Y.; Swanson, K.A.; Muik, A.; Vormehr, M.; Kranz, L.M.; Walzer, K.C.; Hein, S.; Güler, A.; et al. BNT162b Vaccines Protect Rhesus Macaques from SARS-CoV-2. Nature 2021, 592, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-Based Therapeutics--Developing a New Class of Drugs. Nat. Rev Drug Discov 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Lockard, R.E.; Lingrel, J.B. The Synthesis of Mouse Hemoglobin Beta-Chains in a Rabbit Reticulocyte Cell-Free System Programmed with Mouse Reticulocyte 9S RNA. Biochem. Biophys. Res. Commun. 1969, 37, 204–212. [Google Scholar] [CrossRef]
- Panah, B.Y.; Roos, T.; Kunze, M.; Bertsch, F.; Wochner, A.; Rauen, M.; Hoffmann, P. Bioreactor for Rna in Vitro Transcription 2020. U.S. Patent Application No 17/254,853, 28 June 2018. [Google Scholar]
- Skok, J.; Megušar, P.; Vodopivec, T.; Pregeljc, D.; Mencin, N.; Korenč, M.; Krušič, A.; Celjar, A.M.; Pavlin, N.; Krušič, J.; et al. Gram-Scale MRNA Production Using a 250-mL Single-Use Bioreactor. Chem. Ing. Tech. 2022, 94, 1928–1935. [Google Scholar] [CrossRef]
- Ouranidis, A.; Vavilis, T.; Mandala, E.; Davidopoulou, C.; Stamoula, E.; Markopoulou, C.K.; Karagianni, A.; Kachrimanis, K. MRNA Therapeutic Modalities Design, Formulation and Manufacturing under Pharma 4.0 Principles. Biomedicines 2021, 10, 50. [Google Scholar] [CrossRef]
- Ouranidis, A.; Davidopoulou, C.; Tashi, R.-K.; Kachrimanis, K. Pharma 4.0 Continuous MRNA Drug Products Manufacturing. Pharmaceutics 2021, 13, 1371. [Google Scholar] [CrossRef]
- Daniel, S.; Kis, Z.; Kontoravdi, C.; Shah, N. Quality by Design for Enabling RNA Platform Production Processes. Trends Biotechnol. 2022, 40, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K. Making COVID-19 MRNA Vaccines Accessible: Challenges Resolved. Expert. Rev. Vaccines 2022, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Samnuan, K.; Blakney, A.K.; McKay, P.F.; Shattock, R.J. Design-of-Experiments In Vitro Transcription Yield Optimization of Self-Amplifying RNA. Mol. Biol. 2021. [Google Scholar] [CrossRef]
- Tersteeg, S.; Mrozowich, T.; Henrickson, A.; Demeler, B.; Patel, T.R. Purification and Characterization of Inorganic Pyrophosphatase for in Vitro RNA Transcription. Biochem. Cell Biol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Sekhon, S.S.; Shin, W.-R.; Ahn, G.; Cho, B.-K.; Ahn, J.-Y.; Kim, Y.-H. Modifications of MRNA Vaccine Structural Elements for Improving MRNA Stability and Translation Efficiency. Mol. Cell. Toxicol. 2021, 18, 1–8. [Google Scholar] [CrossRef]
- Charenton, C.; Graille, M. MRNA Decapping: Finding the Right Structures. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20180164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; McReynolds, T.; Cabral, C.R.; Escamilla-Powers, J.R.; Houston, M.E. Cap 1 Messenger RNA Synthesis with Co-Transcriptional CleanCap® Analog by In Vitro Transcription. Curr. Protoc. 2021, 1, e39. [Google Scholar] [CrossRef]
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 Vaccine Induces Neutralizing Antibodies and Poly-Specific T Cells in Humans. Nature 2021, 595, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 MRNA Vaccine Design Enabled by Prototype Pathogen Preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring MRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef]
- Cenik, C.; Chua, H.N.; Zhang, H.; Tarnawsky, S.P.; Akef, A.; Derti, A.; Tasan, M.; Moore, M.J.; Palazzo, A.F.; Roth, F.P. Genome Analysis Reveals Interplay between 5′UTR Introns and Nuclear MRNA Export for Secretory and Mitochondrial Genes. PLoS Genet. 2011, 7, e1001366. [Google Scholar] [CrossRef] [PubMed]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated Regions of MRNAs. Genome Biol. 2002, 3, reviews0004.1. [Google Scholar] [CrossRef]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Blagev, D.; Pollack, J.L.; Erle, D.J. Toward a Systematic Understanding of MRNA 3′ Untranslated Regions. Proc. Am. Thorac. Soc. 2011, 8, 163–166. [Google Scholar] [CrossRef] [Green Version]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. MRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving MRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of Antigen-Encoding RNA Increases Stability, Translational Efficacy, and T-Cell Stimulatory Capacity of Dendritic Cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Hia, F.; Takeuchi, O. The Effects of Codon Bias and Optimality on MRNA and Protein Regulation. Cell Mol. Life Sci. 2021, 78, 1909–1928. [Google Scholar] [CrossRef] [PubMed]
- Cannarozzi, G.; Cannarrozzi, G.; Schraudolph, N.N.; Faty, M.; von Rohr, P.; Friberg, M.T.; Roth, A.C.; Gonnet, P.; Gonnet, G.; Barral, Y. A Role for Codon Order in Translation Dynamics. Cell 2010, 141, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Courel, M.; Clément, Y.; Bossevain, C.; Foretek, D.; Vidal Cruchez, O.; Yi, Z.; Bénard, M.; Benassy, M.-N.; Kress, M.; Vindry, C.; et al. GC Content Shapes MRNA Storage and Decay in Human Cells. eLife 2019, 8, e49708. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Keller, J.M.; Weissman, D. Increased Erythropoiesis in Mice Injected with Submicrogram Quantities of Pseudouridine-Containing MRNA Encoding Erythropoietin. Mol. Ther. 2012, 20, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic Acid Agonists for Toll-like Receptor 7 Are Defined by the Presence of Uridine Ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef] [PubMed]
- Minnaert, A.-K.; Vanluchene, H.; Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Raemdonck, K.; Sanders, N.N.; Remaut, K. Strategies for Controlling the Innate Immune Activity of Conventional and Self-Amplifying MRNA Therapeutics: Getting the Message Across. Adv. Drug Deliv. Rev. 2021, 176, 113900. [Google Scholar] [CrossRef] [PubMed]
- Freund, I.; Eigenbrod, T.; Helm, M.; Dalpke, A.H. RNA Modifications Modulate Activation of Innate Toll-Like Receptors. Genes 2019, 10, E92. [Google Scholar] [CrossRef] [Green Version]
- Passmore, L.A.; Coller, J. Roles of MRNA Poly(A) Tails in Regulation of Eukaryotic Gene Expression. Nat. Rev. Mol. Cell. Biol. 2022, 23, 93–106. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.-J. Developing MRNA-Vaccine Technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Jalkanen, A.L.; Coleman, S.J.; Wilusz, J. Determinants and Implications of MRNA Poly(A) Tail Size—Does This Protein Make My Tail Look Big? Semin. Cell Dev. Biol. 2014, 34, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Bardwell, V.J.; Zarkower, D.; Edmonds, M.; Wickens, M. The Enzyme That Adds Poly(A) to MRNAs Is a Classical Poly(A) Polymerase. Mol. Cell. Biol. 1990, 10, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Beverly, M.; Hagen, C.; Slack, O. Poly A Tail Length Analysis of in Vitro Transcribed MRNA by LC-MS. Anal. Bioanal. Chem. 2018, 410, 1667–1677. [Google Scholar] [CrossRef]
- Grier, A.E.; Burleigh, S.; Sahni, J.; Clough, C.A.; Cardot, V.; Choe, D.C.; Krutein, M.C.; Rawlings, D.J.; Jensen, M.C.; Scharenberg, A.M.; et al. PEVL: A Linear Plasmid for Generating MRNA IVT Templates With Extended Encoded Poly(A) Sequences. Mol. Ther. Nucleic Acids 2016, 5, e306. [Google Scholar] [CrossRef]
- To, K.K.W.; Cho, W.C.S. An Overview of Rational Design of MRNA-Based Therapeutics and Vaccines. Expert. Opin. Drug Discov. 2021, 16, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.S.; Prazeres, D.M.F.; Azevedo, A.M.; Marques, M.P.C. MRNA Vaccines Manufacturing: Challenges and Bottlenecks. Vaccine 2021, 39, 2190–2200. [Google Scholar] [CrossRef]
- Sun, B.; Yu, X.; Yin, Y.; Liu, X.; Wu, Y.; Chen, Y.; Zhang, X.; Jiang, C.; Kong, W. Large-Scale Purification of Pharmaceutical-Grade Plasmid DNA Using Tangential Flow Filtration and Multi-Step Chromatography. J. Biosci. Bioeng. 2013, 116, 281–286. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Bulcha, J.T.; Wang, Y.; Ma, H.; Tai, P.W.L.; Gao, G. Viral Vector Platforms within the Gene Therapy Landscape. Sig. Transduct. Target Ther. 2021, 6, 1–24. [Google Scholar] [CrossRef]
- Aliahmad, P.; Miyake-Stoner, S.J.; Geall, A.J.; Wang, N.S. Next Generation Self-Replicating RNA Vectors for Vaccines and Immunotherapies. Cancer Gene Ther. 2022. [Google Scholar] [CrossRef]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zeng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, R.; Agrawal, B. Adenoviral Vector-Based Vaccines and Gene Therapies: Current Status and Future Prospects; IntechOpen: London, UK, 2018; ISBN 978-1-78984-991-2. [Google Scholar]
- Yin, J.; Luan, S. Opportunities and Challenges for the Development of Polymer-Based Biomaterials and Medical Devices. Regen. Biomater. 2016, 3, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Szoka, F.C. Lipid-Based Nanoparticles for Nucleic Acid Delivery. Pharm. Res. 2007, 24, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. MRNA Vaccine Delivery Using Lipid Nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Ryals, R.C.; Patel, S.; Acosta, C.; McKinney, M.; Pennesi, M.E.; Sahay, G. The Effects of PEGylation on LNP Based MRNA Delivery to the Eye. PLoS ONE 2020, 15, e0241006. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Cullis, P.R.; van der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Hu, Y.; Li, A.; Lin, J.; Hsieh, K.; Schneiderman, Z.; Zhang, P.; Zhu, Y.; Qiu, C.; Kokkoli, E.; et al. Payload Distribution and Capacity of MRNA Lipid Nanoparticles. Nat. Commun. 2022, 13, 5561. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tao, W.; Liu, D.; Wu, J.; Guo, Z.; Ji, X.; Bharwani, Z.; Zhao, L.; Zhao, X.; Farokhzad, O.C.; et al. Surface De-PEGylation Controls Nanoparticle-Mediated SiRNA Delivery In Vitro and In Vivo. Theranostics 2017, 7, 1990–2002. [Google Scholar] [CrossRef] [Green Version]
- Eygeris, Y.; Patel, S.; Jozic, A.; Sahay, G. Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery. Nano Lett. 2020, 20, 4543–4549. [Google Scholar] [CrossRef]
- Paunovska, K.; Gil, C.J.; Lokugamage, M.P.; Sago, C.D.; Sato, M.; Lando, G.N.; Gamboa Castro, M.; Bryksin, A.V.; Dahlman, J.E. Analyzing 2000 in Vivo Drug Delivery Data Points Reveals Cholesterol Structure Impacts Nanoparticle Delivery. ACS Nano 2018, 12, 8341–8349. [Google Scholar] [CrossRef] [PubMed]
- Paunovska, K.; Da Silva Sanchez, A.J.; Sago, C.D.; Gan, Z.; Lokugamage, M.P.; Islam, F.Z.; Kalathoor, S.; Krupczak, B.R.; Dahlman, J.E. Nanoparticles Containing Oxidized Cholesterol Deliver MRNA to the Liver Microenvironment at Clinically Relevant Doses. Adv. Mater. 2019, 31, 1807748. [Google Scholar] [CrossRef]
- Carrasco, M.J.; Alishetty, S.; Alameh, M.-G.; Said, H.; Wright, L.; Paige, M.; Soliman, O.; Weissman, D.; Cleveland, T.E.; Grishaev, A.; et al. Ionization and Structural Properties of MRNA Lipid Nanoparticles Influence Expression in Intramuscular and Intravascular Administration. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef]
- Kimura, S.; Khalil, I.A.; Elewa, Y.H.A.; Harashima, H. Novel Lipid Combination for Delivery of Plasmid DNA to Immune Cells in the Spleen. J. Control. Release 2021, 330, 753–764. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, C.; Li, B.; Zhang, X.; Luo, X.; Zeng, C.; Li, W.; Gao, M.; Dong, Y. Lipid Polymer Hybrid Nanomaterials for MRNA Delivery. Cell Mol. Bioeng. 2018, 11, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Analytical Procedures for MRNA Vaccine Quality (Draft Guidelines)|USP-NF. Available online: https://www.uspnf.com/notices/analytical-procedures-mrna-vaccines-20220210 (accessed on 19 August 2022).
- Stöcher, M.; Berg, J. Removal of Template DNA from CRNA Preparations by Combined Oligo (DT) Affinity Chromatography and DNase I Digestion. Biotechniques 2004, 36, 480–482. [Google Scholar] [CrossRef] [Green Version]
- Read, M.L.; Singh, S.; Ahmed, Z.; Stevenson, M.; Briggs, S.S.; Oupicky, D.; Barrett, L.B.; Spice, R.; Kendall, M.; Berry, M.; et al. A Versatile Reducible Polycation-Based System for Efficient Delivery of a Broad Range of Nucleic Acids. Nucleic Acids Res. 2005, 33, e86. [Google Scholar] [CrossRef] [Green Version]
- Gantier, M.P.; Williams, B.R.G. The Response of Mammalian Cells to Double-Stranded RNA. Cytokine Growth Factor Rev. 2007, 18, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Vlatkovic, I.; Ludwig, J.; Boros, G.; Szabó, G.T.; Reichert, J.; Buff, M.; Baiersdörfer, M.; Reinholz, J.; Mahiny, A.J.; Şahin, U.; et al. Ribozyme Assays to Quantify the Capping Efficiency of In Vitro-Transcribed MRNA. Pharmaceutics 2022, 14, 328. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D.J.A.; Volkin, D.B.; Hoogendoorn, K.H.; Lubiniecki, A.S.; Jiskoot, W. The Science Is There: Key Considerations for Stabilizing Viral Vector-Based Covid-19 Vaccines. JPharmSci. 2021, 110, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Crommelin, D.J.A.; Anchordoquy, T.J.; Volkin, D.B.; Jiskoot, W.; Mastrobattista, E. Addressing the Cold Reality of MRNA Vaccine Stability. J. Pharm. Sci. 2021, 110, 997–1001. [Google Scholar] [CrossRef]
- Mitchell, J.J.; Trakadis, Y.J.; Scriver, C.R. Phenylalanine Hydroxylase Deficiency. Genet. Med.: Off. J. Am. Coll. Med. Genet. 2011, 13, 697–707. [Google Scholar] [CrossRef]
- Lichter-Konecki, U.; Vockley, J. Phenylketonuria: Current Treatments and Future Developments. Drugs 2019, 79, 495–500. [Google Scholar] [CrossRef]
- Blau, N. Defining Tetrahydrobiopterin (BH4)-Responsiveness in PKU. J. Inherit. Metab. Dis. 2008, 31, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Blau, N.; Bélanger-Quintana, A.; Demirkol, M.; Feillet, F.; Giovannini, M.; MacDonald, A.; Trefz, F.K.; van Spronsen, F.J. Optimizing the Use of Sapropterin (BH(4)) in the Management of Phenylketonuria. Mol. Genet. Metab. 2009, 96, 158–163. [Google Scholar] [CrossRef]
- Perez-Garcia, C.G.; Diaz-Trelles, R.; Vega, J.B.; Bao, Y.; Sablad, M.; Limphong, P.; Chikamatsu, S.; Yu, H.; Taylor, W.; Karmali, P.P.; et al. Development of an MRNA Replacement Therapy for Phenylketonuria. Mol. Therapy. Nucleic Acids 2022, 28, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Truong, B.; Allegri, G.; Liu, X.-B.; Burke, K.E.; Zhu, X.; Cederbaum, S.D.; Häberle, J.; Martini, P.G.V.; Lipshutz, G.S. Lipid Nanoparticle-Targeted MRNA Therapy as a Treatment for the Inherited Metabolic Liver Disorder Arginase Deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 21150–21159. [Google Scholar] [CrossRef] [PubMed]
- Sin, Y.Y.; Baron, G.; Schulze, A.; Funk, C.D. Arginase-1 Deficiency. J. Mol. Med. 2015, 93, 1287–1296. [Google Scholar] [CrossRef]
- Braga, A.C.; Vilarinho, L.; Ferreira, E.; Rocha, H. Hyperargininemia Presenting as Persistent Neonatal Jaundice and Hepatic Cirrhosis. J. Pediatr. Gastroenterol. Nutr. 1997, 24, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, F.; Lee, B. Clinical, Biochemical, and Molecular Spectrum of Hyperargininemia Due to Arginase I Deficiency. Am. J. Med. Genetics. Part C Semin. Med. Genet. 2006, 142C, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandler, R.J. Messenger RNA Therapy as an Option for Treating Metabolic Disorders. Proc. Natl. Acad. Sci. USA 2019, 116, 20804–20806. [Google Scholar] [CrossRef] [Green Version]
- DeRosa, F.; Smith, L.; Shen, Y.; Huang, Y.; Pan, J.; Xie, H.; Yahalom, B.; Heartlein, M.W. Improved Efficacy in a Fabry Disease Model Using a Systemic MRNA Liver Depot System as Compared to Enzyme Replacement Therapy. Mol. Ther. 2019, 27, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of MRNA Therapeutics for the Treatment of Hepatic Diseases. Mol. Ther.: J. Am. Soc. Gene Ther. 2019, 27, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Berraondo, P.; Martini, P.G.V.; Avila, M.A.; Fontanellas, A. Messenger RNA Therapy for Rare Genetic Metabolic Diseases. Gut 2019, 68, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Russick, J.; Delignat, S.; Milanov, P.; Christophe, O.; Boros, G.; Denis, C.V.; Lenting, P.J.; Kaveri, S.V.; Lacroix-Desmazes, S. Correction of Bleeding in Experimental Severe Hemophilia A by Systemic Delivery of Factor VIII-Encoding MRNA. Haematologica 2020, 105, 1129–1137. [Google Scholar] [CrossRef]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic Delivery of Factor IX Messenger RNA for Protein Replacement Therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified MRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [Green Version]
- Haque, A.K.M.A.; Dewerth, A.; Antony, J.S.; Riethmüller, J.; Schweizer, G.R.; Weinmann, P.; Latifi, N.; Yasar, H.; Pedemonte, N.; Sondo, E.; et al. Chemically Modified HCFTR MRNAs Recuperate Lung Function in a Mouse Model of Cystic Fibrosis. Sci. Rep. 2018, 8, 16776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, T.M.; Konstan, M.W. What Is Cystic Fibrosis? JAMA 2022, 327, 191. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Sanchez, A.; Paunovska, K.; Cristian, A.; Dahlman, J.E. Treating Cystic Fibrosis with MRNA and CRISPR. Hum. Gene Ther. 2020, 31, 940–955. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; Del Pozo-Rodríguez, A. Nanomedicines to Deliver MRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, A.S.; Beck, S.; Meyer, M.; Penque, D.; Cutting, G.R.; Amaral, M.D. Five Percent of Normal Cystic Fibrosis Transmembrane Conductance Regulator MRNA Ameliorates the Severity of Pulmonary Disease in Cystic Fibrosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anttila, V.; Saraste, A.; Knuuti, J.; Jaakkola, P.; Hedman, M.; Svedlund, S.; Lagerström-Fermér, M.; Kjaer, M.; Jeppsson, A.; Gan, L.-M. Synthetic MRNA Encoding VEGF-A in Patients Undergoing Coronary Artery Bypass Grafting: Design of a Phase 2a Clinical Trial. Mol. Ther. Methods Clin. Dev. 2020, 18, 464–472. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. A Randomized, Double-Blind, Placebo-Controlled, Multi-Centre, Sequential Design, Phase IIa Study to Evaluate Safety and Tolerability of Epicardial Injections of AZD8601 During Coronary Artery Bypass Grafting Surgery; clinicaltrials.gov, 2021. Available online: https://clinicaltrials.gov/ct2/show/study/NCT03370887 (accessed on 9 November 2022).
- Sun, N.; Ning, B.; Hansson, K.M.; Bruce, A.C.; Seaman, S.A.; Zhang, C.; Rikard, M.; DeRosa, C.A.; Fraser, C.L.; Wågberg, M.; et al. Modified VEGF-A MRNA Induces Sustained Multifaceted Microvascular Response and Accelerates Diabetic Wound Healing. Sci. Rep. 2018, 8, 17509. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, L.; Clarke, J.C.; Yen, C.; Gregoire, F.; Albery, T.; Billger, M.; Egnell, A.-C.; Gan, L.-M.; Jennbacken, K.; Johansson, E.; et al. Biocompatible, Purified VEGF-A MRNA Improves Cardiac Function after Intracardiac Injection 1 Week Post-Myocardial Infarction in Swine. Mol. Ther. Methods Clin. Dev. 2018, 9, 330–346. [Google Scholar] [CrossRef] [PubMed]
- VEGF (Human). Available online: https://www.phosphosite.org/proteinAction.action?id=25543000&showAllSites=true (accessed on 21 December 2022).
- Lam Hon Wah, A.M.; Lam, K.F.; Tsui, F.; Robinson, B.; Saunders, M.E.; Gravel, R.A. Assignment of the Alpha and Beta Chains of Human Propionyl-CoA Carboxylase to Genetic Complementation Groups. Am. J. Hum. Genet. 1983, 35, 889–899. [Google Scholar] [PubMed]
- Chandler, R.J.; Tsai, M.S.; Dorko, K.; Sloan, J.; Korson, M.; Freeman, R.; Strom, S.; Venditti, C.P. Adenoviral-Mediated Correction of Methylmalonyl-CoA Mutase Deficiency in Murine Fibroblasts and Human Hepatocytes. BMC Med. Genet. 2007, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Caldovic, L.; Abdikarim, I.; Narain, S.; Tuchman, M.; Morizono, H. Genotype–Phenotype Correlations in Ornithine Transcarbamylase Deficiency: A Mutation Update. J. Genet. Genom. 2015, 42, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Riordan, C.R.; Lachapelle, A.L.; Marshall, J.; Higgins, E.A.; Cheng, S.H. Characterization of the Oligosaccharide Structures Associated with the Cystic Fibrosis Transmembrane Conductance Regulator. Glycobiology 2000, 10, 1225–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateus, J.; Dan, J.M.; Zhang, Z.; Rydyznski Moderbacher, C.; Lammers, M.; Goodwin, B.; Sette, A.; Crotty, S.; Weiskopf, D. Low-Dose MRNA-1273 COVID-19 Vaccine Generates Durable Memory Enhanced by Cross-Reactive T Cells. Science 2021, 374, eabj9853. [Google Scholar] [CrossRef]
- Translate Bio, Inc. A Phase 1/2, Randomized, Double-Blinded, Placebo-Controlled, Combined Single and Multiple Ascending Dose Study Evaluating the Safety, Tolerability, and Biological Activity of MRT5005 Administered by Nebulization to Adult Subjects With Cystic Fibrosis. Available online: https://clinicaltrials.gov/ct2/show/NCT03375047 (accessed on 20 December 2022).
- Uddin, M.N.; Roni, M.A. Challenges of Storage and Stability of MRNA-Based COVID-19 Vaccines. Vaccines 2021, 9, 1033. [Google Scholar] [CrossRef]
- Zhang, M.; Hussain, A.; Yang, H.; Zhang, J.; Liang, X.-J.; Huang, Y. MRNA-Based Modalities for Infectious Disease Management. Nano Res. 2022. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, X.; Yan, J.; Du, S.; Xue, Y.; Li, W.; Xiang, G.; Dong, Y. Long-Term Storage of Lipid-like Nanoparticles for MRNA Delivery. Bioact. Mater. 2020, 5, 358–363. [Google Scholar] [CrossRef]
- Muramatsu, H.; Lam, K.; Bajusz, C.; Laczkó, D.; Karikó, K.; Schreiner, P.; Martin, A.; Lutwyche, P.; Heyes, J.; Pardi, N. Lyophilization Provides Long-Term Stability for a Lipid Nanoparticle-Formulated, Nucleoside-Modified MRNA Vaccine. Mol. Ther. 2022, 30, 1941–1951. [Google Scholar] [CrossRef]
- Huang, X.; Kong, N.; Zhang, X.; Cao, Y.; Langer, R.; Tao, W. The Landscape of MRNA Nanomedicine. Nat. Med. 2022, 28, 2273–2287. [Google Scholar] [CrossRef] [PubMed]
- Papukashvili, D.; Rcheulishvili, N.; Liu, C.; Ji, Y.; He, Y.; Wang, P.G. Self-Amplifying RNA Approach for Protein Replacement Therapy. Int. J. Mol. Sci. 2022, 23, 12884. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and Its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenstra, T.D.; Pauley, B.; Injeti, E.; Rotello, R.J. In Vitro Characterization of SARS-CoV-2 Protein Translated from the Moderna MRNA-1273 Vaccine. Allergy Immunol. 2022. [Google Scholar] [CrossRef]
- Davis, J.; Debear, J.; Cheng, C. Polynucleotide Purification with Monolith Columns 2017. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017205477 (accessed on 16 October 2022).
- Geiger, J.; Treml, M. Mrna Purification by Tangential Flow Filtration 2020. Available online: https://patents.google.com/patent/WO2020165158A1/en (accessed on 15 October 2022).
- Chatterjee, A.; Mirer, P.L.; Zaldivar Santamaria, E.; Klapperich, C.; Sharon, A.; Sauer-Budge, A.F. RNA Isolation from Mammalian Cells Using Porous Polymer Monoliths: An Approach for High-Throughput Automation. Anal. Chem. 2010, 82, 4344–4356. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT NUMBER/PHASE | CONDITION | DELIVERY SYSTEM | ENCODING SEQUENCE/PROTEIN MOLECULAR WEIGHT | SUBJECTS | INTERVENTION | STATUS |
|---|---|---|---|---|---|---|
| NCT03370887/Phase II | Heart failure | Naked mRNA | VEGF-A 27 kDa [126] | Twenty-four patients with compromised left ventricular function that undergo surgical revascularization. | Patients had received either AZD8601 or placebo as epicardial injections and were followed up for six months. | Completed |
| NCT02935712/Phase I | Male subjects with type II diabetes | Naked mRNA | VEGF-A 27 kDa [126] | Up to sixty male patients with type II diabetes, aged 18–65 years old | In Part A, subjects had received an intradermal injection (ID) of either AZD8601 or placebo in a single ascending dose. In Part B, patients had received an ID injection of either AZD8601, in forearm skin, or the placebo. | Completed |
| NCT04159103/Phase I/II | Propionic Acidemia (PA) | LNPs | Alpha and Beta subunits of propionyl-CoA carboxylaseAlpha chain: 72 kDa Beta chain: 56 kDA [127] | Thirty-six patients with genetically confirmed PA, from one year old and older. | In Phase I, the patients will receive doses of mRNA-3927, for the dose optimization stage and subsequently for the dose expansion stage. In Phase II, the patients will receive the identified intravenous dose of mRNA-3927 and will be followed up for two years. | Recruiting |
| NCT03810690/Phase I/II | Methylmalonic acidemia (MMA) | LNPs | Methylmalonyl-coenzyme A mutase (MUT) 78 kDa [128] | Patients with methylmalonic academia, aged 1–18 years old, with elevated plasma methylmalonic acid. | The patients were about to receive doses of mRNA-3704, for the dose escalation phase, and subsequently for the dose expansion stage. | Withdrawn |
| NCT03767270/Phase I/II | Ornithine transcarbamylase deficiency (OTCD) | LNPs | Ornithine transcarbamylase 36.1 kDa [129] | Subjects with OTC Deficiency | The patients were about to receive intravenous, single-ascending low, mid and high doses of MRT5201 or the placebo. | Withdrawn |
| NCT03375047/Phase I/II | Cystic fibrosis (CF) | LNPs | Human cystic fibrosis transmembrane regulator protein (CFTR) 127 kDa [130] | Forty adult subjects with CF | The patients are supposed to receive single and multiple escalating doses of MRT5005, administered by nebulization to the respiratory tract, or the placebo. | Unknown |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vavilis, T.; Stamoula, E.; Ainatzoglou, A.; Sachinidis, A.; Lamprinou, M.; Dardalas, I.; Vizirianakis, I.S. mRNA in the Context of Protein Replacement Therapy. Pharmaceutics 2023, 15, 166. https://doi.org/10.3390/pharmaceutics15010166
Vavilis T, Stamoula E, Ainatzoglou A, Sachinidis A, Lamprinou M, Dardalas I, Vizirianakis IS. mRNA in the Context of Protein Replacement Therapy. Pharmaceutics. 2023; 15(1):166. https://doi.org/10.3390/pharmaceutics15010166
Chicago/Turabian StyleVavilis, Theofanis, Eleni Stamoula, Alexandra Ainatzoglou, Athanasios Sachinidis, Malamatenia Lamprinou, Ioannis Dardalas, and Ioannis S. Vizirianakis. 2023. "mRNA in the Context of Protein Replacement Therapy" Pharmaceutics 15, no. 1: 166. https://doi.org/10.3390/pharmaceutics15010166